
CLASH Analyst: A Web Server to Identify In Vivo RNA–RNA Interactions from CLASH Data

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
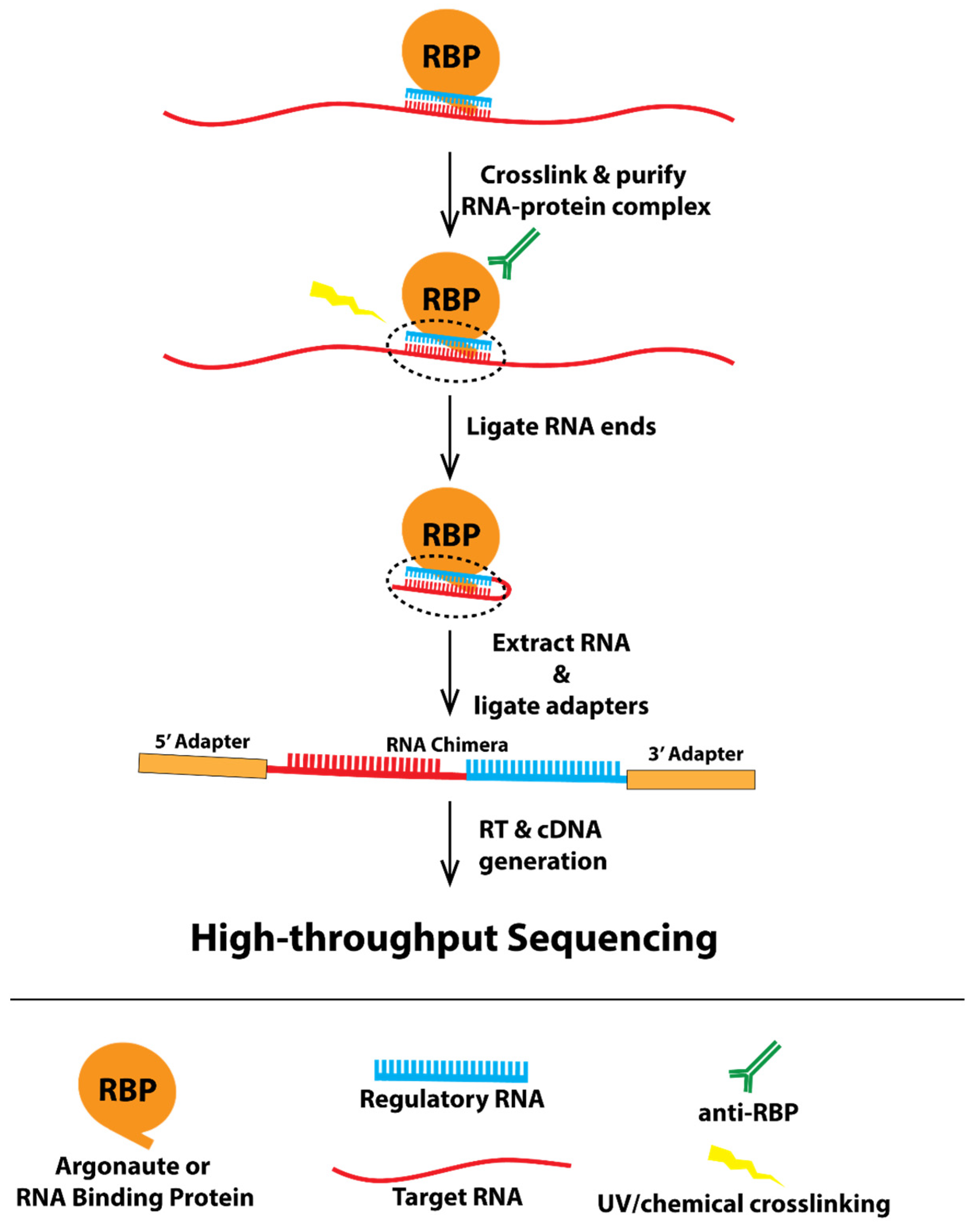
:1. Introduction
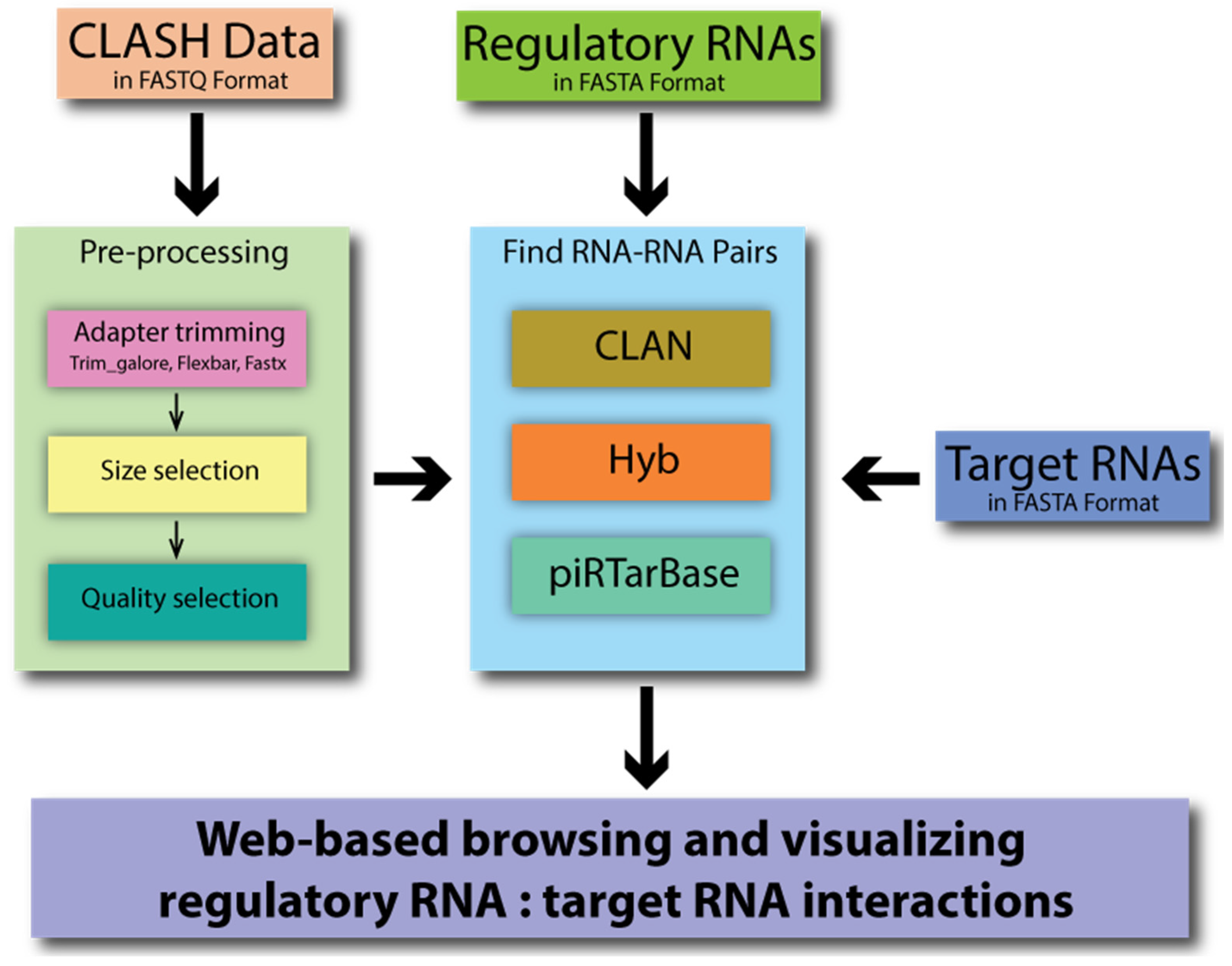
2. Webtool Description
2.1. General Framework
2.2. Data Input
2.3. CLASH Read Preprocessing
2.4. Identify Chimeras
2.5. User Information
2.6. Chimeric Reads Output
2.6.1. Browse by Regulatory RNA Name
2.6.2. Browse by Target RNA Name
2.6.3. Browse by RNA–RNA Pair
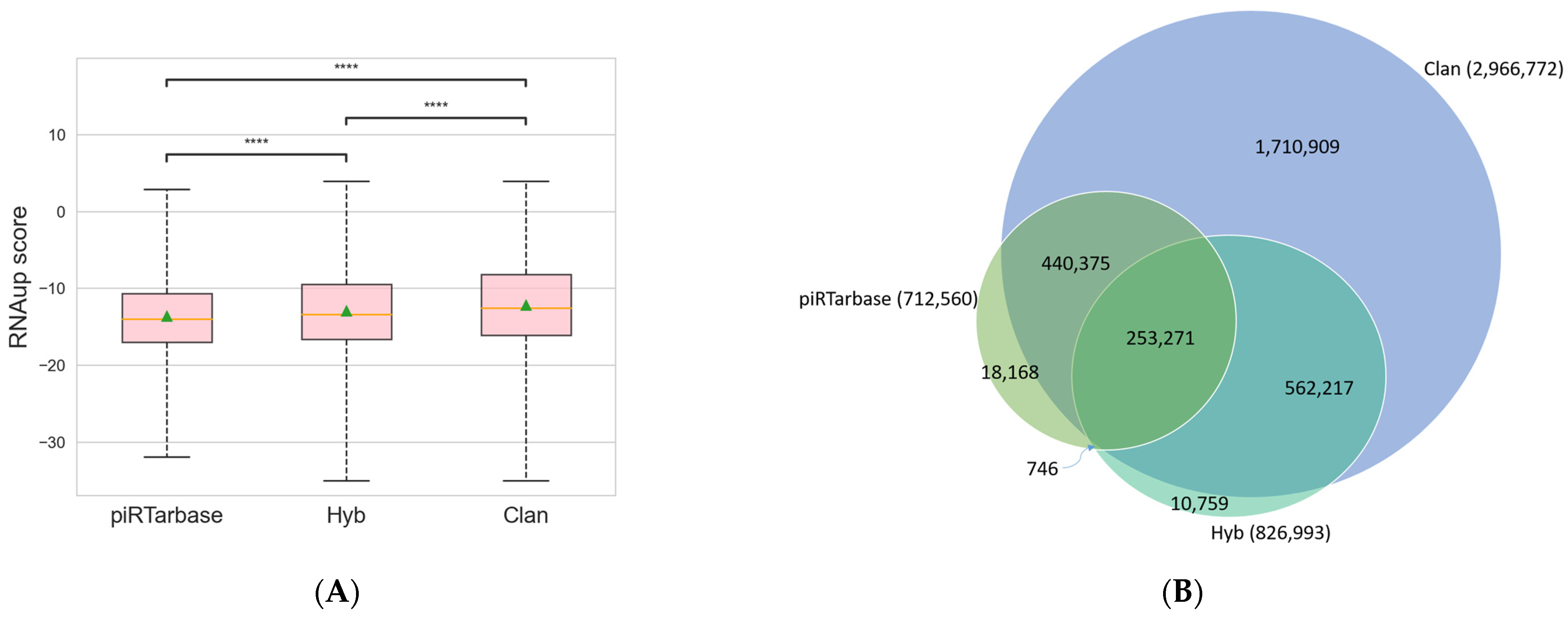
3. Analysis Results
4. Discussion
5. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudla, G.; Granneman, S.; Hahn, D.; Beggs, J.D.; Tollervey, D. Cross-linking, ligation, and sequencing of hybrids reveals RNA-RNA interactions in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 10010–10015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the Human miRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Shen, E.-Z.; Chen, H.; Ozturk, A.R.; Tu, S.; Shirayama, M.; Tang, W.; Ding, Y.-H.; Dai, S.-Y.; Weng, Z.; Mello, C.C. Identification of piRNA Binding Sites Reveals the Argonaute Regulatory Landscape of the C. elegans Germline. Cell 2018, 172, 937–951.e18. [Google Scholar] [CrossRef] [Green Version]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP Identifies Nova-Regulated RNA Networks in the Brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Scheel, T.K.H.; Luna, J.M.; Park, C.Y.; Fak, J.J.; Nishiuchi, E.; Rice, C.M.; Darnell, R.B. MiRNA-target chimeras reveal miRNA 3′-end pairing as a major determinant of Argonaute target specificity. Nat. Commun. 2015, 6, 8864. [Google Scholar] [CrossRef]
- Zhong, C.; Zhang, S. Accurate and Efficient Mapping of the Cross-Linked microRNA-mRNA Duplex Reads. iScience 2019, 18, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, A.J.; Moody, J.; Helwak, A.; Tollervey, D.; Kudla, G. Hyb: A bioinformatics pipeline for the analysis of CLASH (crosslinking, ligation and sequencing of hybrids) data. Methods 2014, 65, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Videm, P.; Kumar, A.; Zharkov, O.; Grüning, B.A.; Backofen, R. ChiRA: An integrated framework for chimeric read analysis from RNA-RNA interactome and RNA structurome data. Gigascience 2021, 10, giaa158. [Google Scholar] [CrossRef]
- Blankenberg, D.; Coraor, N.; Von Kuster, G.; Taylor, J.; Nekrutenko, A. Integrating diverse databases into an unified analysis framework: A Galaxy approach. Database 2011, 2011, bar011. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.S.; Brown, J.S.; Chen, T.T.; Chu, Y.H.; Huang, W.C.; Tu, S.; Lee, H.C. PiRTarBase: A database of piRNA targeting sites and their roles in gene regulation. Nucleic Acids Res. 2019, 47, D181–D187. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Dodt, M.; Roehr, J.T.; Ahmed, R.; Dieterich, C. FLEXBAR-flexible barcode and adapter processing for next-generation sequencing platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef] [Green Version]
- H. Lab, “FASTX-Toolkit (RRID:SCR_005534)”. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 6 January 2022).
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 1–14. [Google Scholar] [CrossRef]
- Shigematsu, M.; Kirino, Y. tRNA-derived short non-coding RNA as interacting partners of argonaute proteins. Gene Regul. Syst. Biol. 2015, 9, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.C.; Cao, X.; Yu, P.; Xiao, S.; Lu, J.; Biase, F.H.; Sridhar, B.; Huang, N.; Zhang, K.; Zhong, S. Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nat. Commun. 2016, 7, 12023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, C.; Cai, Z.; Ye, R.; Su, R.; Hu, N.; Zhao, H.; Xue, Y. Global in situ profiling of RNA-RNA spatial interactions with RIC-seq. Nat. Protoc. 2021, 16, 2916–2946. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.-S.; Brown, J.S.; Chen, P.-H.; Shiue, S.-C.; Lee, D.-E.; Lee, H.-C. CLASH Analyst: A Web Server to Identify In Vivo RNA–RNA Interactions from CLASH Data. Non-Coding RNA 2022, 8, 6. https://doi.org/10.3390/ncrna8010006
Wu W-S, Brown JS, Chen P-H, Shiue S-C, Lee D-E, Lee H-C. CLASH Analyst: A Web Server to Identify In Vivo RNA–RNA Interactions from CLASH Data. Non-Coding RNA. 2022; 8(1):6. https://doi.org/10.3390/ncrna8010006
Chicago/Turabian StyleWu, Wei-Sheng, Jordan S. Brown, Pin-Hao Chen, Sheng-Cian Shiue, Dong-En Lee, and Heng-Chi Lee. 2022. "CLASH Analyst: A Web Server to Identify In Vivo RNA–RNA Interactions from CLASH Data" Non-Coding RNA 8, no. 1: 6. https://doi.org/10.3390/ncrna8010006
APA StyleWu, W. -S., Brown, J. S., Chen, P. -H., Shiue, S. -C., Lee, D. -E., & Lee, H. -C. (2022). CLASH Analyst: A Web Server to Identify In Vivo RNA–RNA Interactions from CLASH Data. Non-Coding RNA, 8(1), 6. https://doi.org/10.3390/ncrna8010006