
LncRNAs Are Differentially Expressed between Wildtype and Cell Line Strains of African Trypanosomes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
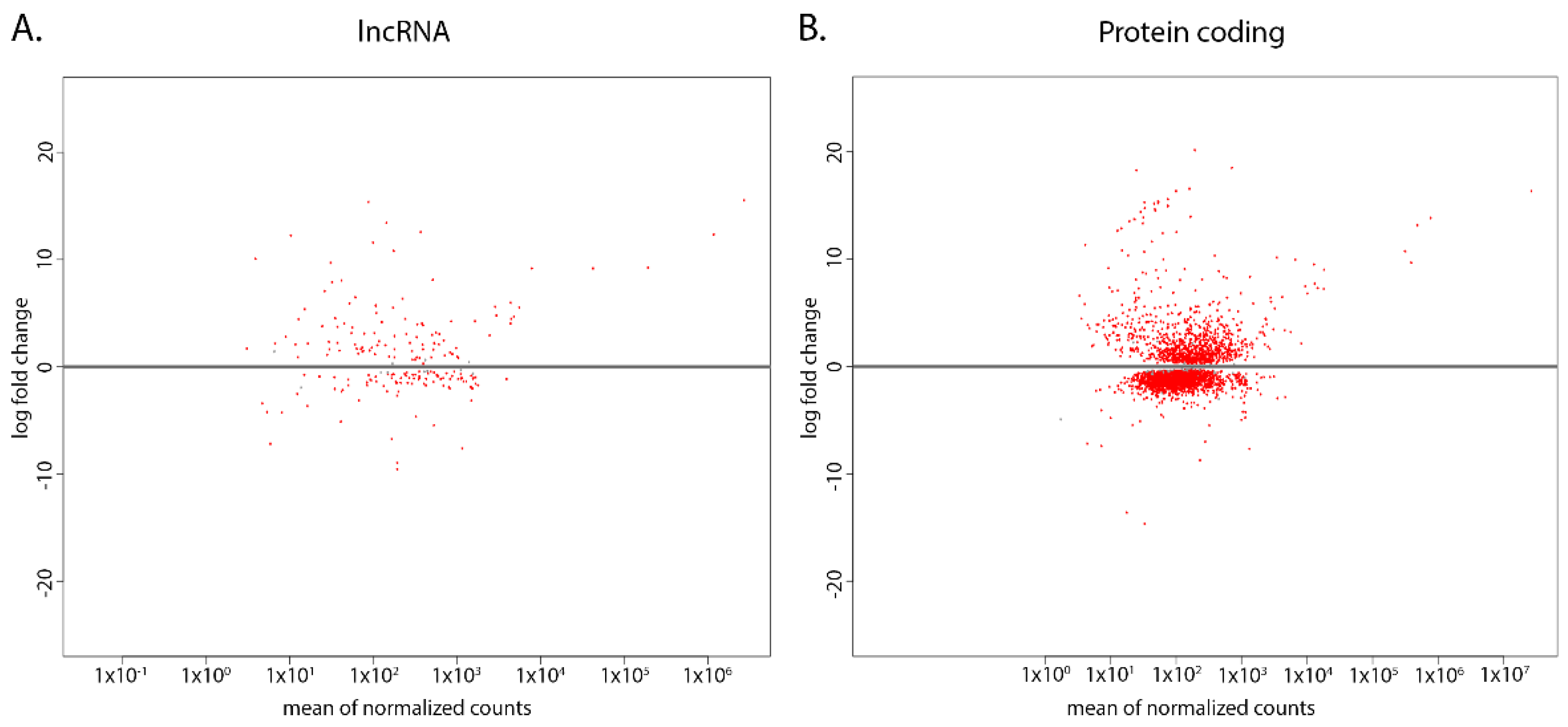
2.1. Novel lncRNAs Were Identified in Both CL and WT T. brucei
2.1.1. CL and WT T. brucei lncRNA Expression Patterns Are Distinct
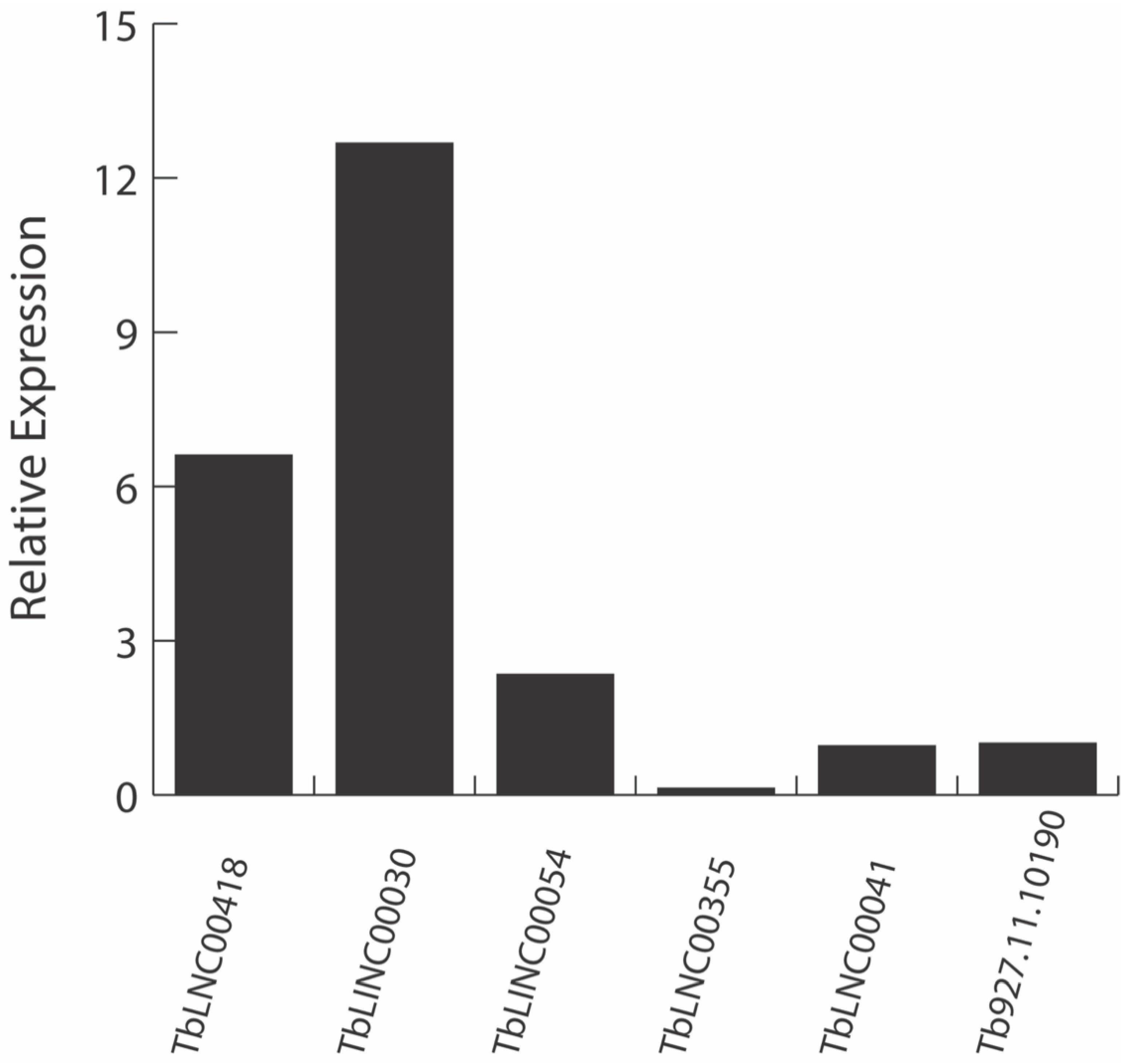
2.1.2. RT-qPCR Assay Confirms the In Silico lncRNA Expression in T. brucei
3. Discussion
4. Materials and Methods
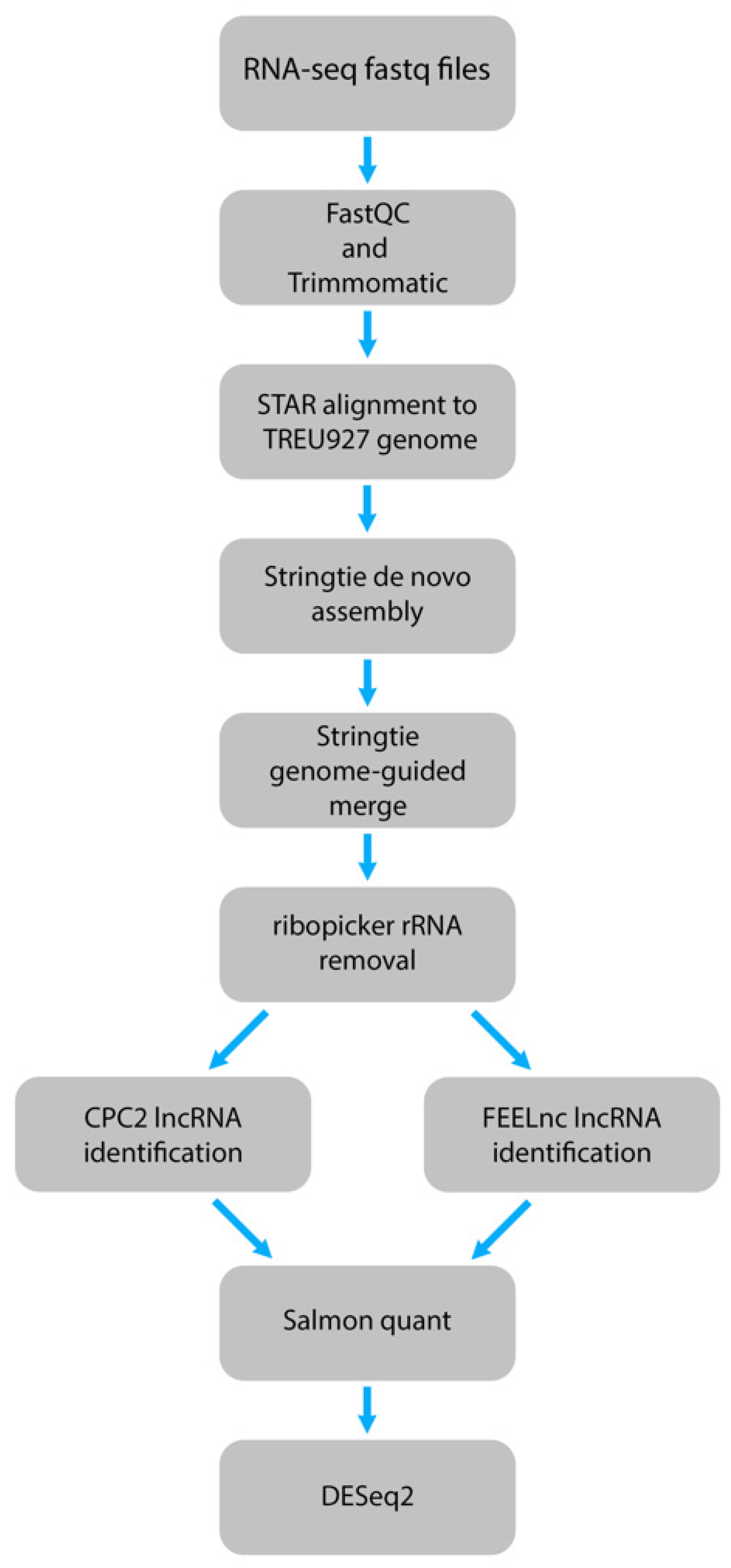
4.1. Genomic and Transcriptomic Data Analysis
4.2. Identification of Long Noncoding RNAs
4.3. Statistical Analyses
4.4. Animals and Parasites
4.5. RNA Extraction and cDNA Generation
4.6. RT-qPCR Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, P.G.E. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013, 12, 186–194. [Google Scholar] [CrossRef]
- Pays, E.; Vanhollebeke, B. Human innate immunity against African trypanosomes. Curr. Opin. Immunol. 2009, 21, 493–498. [Google Scholar] [CrossRef] [PubMed]
- De Greef, C.; Imberechts, H.; Matthyssens, G.; Van Meirvenne, N.; Hamers, R. A gene expressed only in serum-resistant variants of Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol. 1989, 36, 169–176. [Google Scholar] [CrossRef]
- Van Xong, H.; Vanhamme, L.; Chamekh, M.; Chimfwembe, C.E.; Van Den Abbeele, J.; Pays, A.; Van Meirvenne, N.; Hamers, R.; De Baetselier, P.; Pays, E. A VSG Expression Site–Associated Gene Confers Resistance to Human Serum in Trypanosoma rhodesiense. Cell 1998, 95, 839–846. [Google Scholar] [CrossRef] [Green Version]
- Capewell, P.; Clucas, C.; DeJesus, E.; Kieft, R.; Hajduk, S.; Veitch, N.; Steketee, P.C.; Cooper, A.; Weir, W.; MacLeod, A. The TgsGP gene is essential for resistance to human serum in Trypanosoma brucei gambiense. PLoS Pathog. 2013, 9, e1003686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capewell, P.; Veitch, N.J.; Turner, C.M.R.; Raper, J.; Berriman, M.; Hajduk, S.L.; MacLeod, A. Differences between Trypanosoma brucei gambiense groups 1 and 2 in their resistance to killing by trypanolytic factor 1. PLoS Negl. Trop. Dis. 2011, 5, e1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJesus, E.; Kieft, R.; Albright, B.; Stephens, N.A.; Hajduk, S.L. A single amino acid substitution in the group 1 Trypanosoma brucei gambiense haptoglobin-hemoglobin receptor abolishes TLF-1 binding. PLoS Pathog. 2013, 9, e1003317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieft, R.; Capewell, P.; Turner, C.M.R.; Veitch, N.J.; MacLeod, A.; Hajduk, S. Mechanism of Trypanosoma brucei gambiense (group 1) resistance to human trypanosome lytic factor. Proc. Natl. Acad. Sci. USA 2010, 107, 16137–16141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzureau, P.; Uzureau, S.; Lecordier, L.; Fontaine, F.; Tebabi, P.; Homblé, F.; Grélard, A.; Zhendre, V.; Nolan, D.P.; Lins, L.; et al. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature 2013, 501, 430–434. [Google Scholar] [CrossRef]
- Sistrom, M.; Evans, B.; Benoit, J.; Balmer, O.; Aksoy, S.; Caccone, A. De Novo Genome Assembly Shows Genome Wide Similarity between Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense. PLoS ONE 2016, 11, e0147660. [Google Scholar] [CrossRef] [Green Version]
- Baldauf, S.L.; Roger, A.J.; Wenk-Siefert, I.; Doolittle, W.F. A Kingdom-Level Phylogeny of Eukaryotes Based on Combined Protein Data. Science 2000, 290, 972. [Google Scholar] [CrossRef]
- Hirumi, H.; Doyle, J.J.; Hirumi, K. African trypanosomes: Cultivation of animal-infective Trypanosoma brucei in vitro. Science 1977, 196, 992. [Google Scholar] [CrossRef]
- Hirumi, H.; Hirumi, K. Continuous Cultivation of Trypanosoma brucei Blood Stream Forms in a Medium Containing a Low Concentration of Serum Protein without Feeder Cell Layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef] [Green Version]
- Gibson, W. The origins of the trypanosome genome strains Trypanosoma brucei brucei TREU 927, T. b. gambiense DAL 972, T. vivax Y486 and T. congolense IL3000. Parasites Vectors 2012, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu Daniella, C.; Lennard Nicola, J.; Caler, E.; Hamlin Nancy, E.; Haas, B.; et al. The Genome of the African Trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Januszyk, M.; Rennert, R.C.; Sorkin, M.; Maan, Z.N.; Wong, L.K.; Whittam, A.J.; Whitmore, A.; Duscher, D.; Gurtner, G.C. Evaluating the Effect of Cell Culture on Gene Expression in Primary Tissue Samples Using Microfluidic-Based Single Cell Transcriptional Analysis. Microarrays 2015, 4, 540–550. [Google Scholar] [CrossRef]
- Zaitseva, M.; Vollenhoven, B.J.; Rogers, P.A.W. In vitro culture significantly alters gene expression profiles and reduces differences between myometrial and fibroid smooth muscle cells. Mol. Hum. Reprod. 2006, 12, 187–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, L.E.; Koslowsky, D. Cell-line specific RNA editing patterns in Trypanosoma brucei suggest a unique mechanism to generate protein variation in a system intolerant to genetic mutations. Nucleic Acids Res. 2020, 48, 1479–1493. [Google Scholar] [CrossRef]
- Mulindwa, J.; Leiss, K.; Ibberson, D.; Kamanyi Marucha, K.; Helbig, C.; Melo do Nascimento, L.; Silvester, E.; Matthews, K.; Matovu, E.; Enyaru, J.; et al. Transcriptomes of Trypanosoma brucei rhodesiense from sleeping sickness patients, rodents and culture: Effects of strain, growth conditions and RNA preparation methods. PLoS Negl. Trop. Dis. 2018, 12, e0006280. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermüller, J.; Hofacker, I.L.; et al. RNA Maps Reveal New RNA Classes and a Possible Function for Pervasive Transcription. Science 2007, 316, 1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Chainani, P.; White, T.; Yang, J.; Liu, Y.; Soibam, B. Deep learning identifies genome-wide DNA binding sites of long non-coding RNAs. RNA Biol. 2018, 15, 1468–1476. [Google Scholar] [CrossRef]
- Jadaliha, M.; Gholamalamdari, O.; Tang, W.; Zhang, Y.; Petracovici, A.; Hao, Q.; Tariq, A.; Kim, T.G.; Holton, S.E.; Singh, D.K.; et al. A natural antisense lncRNA controls breast cancer progression by promoting tumor suppressor gene mRNA stability. PLoS Genet. 2018, 14, e1007802. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by non-coding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic non-coding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic non-coding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beletskii, A.; Hong, Y.K.; Pehrson, J.; Egholm, M.; Strauss, W.M. PNA interference mapping demonstrates functional domains in the non-coding RNA Xist. Proc. Natl. Acad. Sci. USA 2001, 98, 9215–9220. [Google Scholar] [CrossRef] [Green Version]
- Saghafi, T.; Taheri, R.A.; Parkkila, S.; Emameh, R.Z. Phytochemicals as Modulators of Long Non-Coding RNAs and Inhibitors of Cancer-Related Carbonic Anhydrases. Int. J. Mol. Sci. 2019, 20, 2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guegan, F.; Bento, F.; Neves, D.; Sequeira, M.; Notredame, C.; Figueiredo, L.M. A long non-coding RNA controls parasite differentiation in African trypanosomes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Jiao, F.; Fang, S.; Tan, C.; Liu, Z.; Zhao, Y. Evolutionary annotation of conserved long non-coding RNAs in major mammalian species. Sci. China Life Sci. 2015, 58, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Necsulea, A.; Soumillon, M.; Warnefors, M.; Liechti, A.; Daish, T.; Zeller, U.; Baker, J.C.; Grützner, F.; Kaessmann, H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635–640. [Google Scholar] [CrossRef]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long non-coding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Naguleswaran, A.; Doiron, N.; Roditi, I. RNA-Seq analysis validates the use of culture-derived Trypanosoma brucei and provides new markers for mammalian and insect life-cycle stages. BMC Genom. 2018, 19, 227. [Google Scholar] [CrossRef] [Green Version]
- Trindade, S.; Rijo-Ferreira, F.; Carvalho, T.; Pinto-Neves, D.; Guegan, F.; Aresta-Branco, F.; Bento, F.; Young, S.A.; Pinto, A.; Van Den Abbeele, J.; et al. Trypanosoma brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell Host Microbe 2016, 19, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Jensen, B.C.; Ramasamy, G.; Vasconcelos, E.J.R.; Ingolia, N.T.; Myler, P.J.; Parsons, M. Extensive stage-regulation of translation revealed by ribosome profiling of Trypanosoma brucei. BMC Genom. 2014, 15, 911. [Google Scholar] [CrossRef] [Green Version]
- Kolev, N.G.; Franklin, J.B.; Carmi, S.; Shi, H.; Michaeli, S.; Tschudi, C. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010, 6, e1001090. [Google Scholar] [CrossRef] [Green Version]
- Wucher, V.; Legeai, F.; Hédan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanamamalai, V.K.; Garg, P.; Kolluri, G.; Gandham, R.K.; Jali, I.; Sharma, S. Transcriptomic analysis to infer key molecular players involved during host response to NDV challenge in Gallus gallus (Leghorn & Fayoumi). Sci. Rep. 2021, 11, 8486. [Google Scholar] [PubMed]
- Xue, J.; Lv, Q.; Khas, E.; Bai, C.; Ma, B.; Li, W.; Cao, Q.; Fan, Z.; Ao, C. Tissue-specific regulatory mechanism of LncRNAs and methylation in sheep adipose and muscle induced by Allium mongolicum Regel extracts. Sci. Rep. 2021, 11, 9186. [Google Scholar] [CrossRef] [PubMed]
- Salabi, F.; Jafari, H.; Navidpour, S.; Sadr, A.S. Systematic and computational identification of Androctonus crassicauda long non-coding RNAs. Sci. Rep. 2021, 11, 4720. [Google Scholar] [CrossRef]
- Kim, H.C.; Khalil, A.M.; Jolly, E.R. LncRNAs in molluscan and mammalian stages of parasitic schistosomes are developmentally-regulated and coordinately expressed with protein-coding genes. RNA Biol. 2020, 17, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Akay, A.; Jordan, D.; Navarro, I.C.; Wrzesinski, T.; Ponting, C.P.; Miska, E.A.; Haerty, W. Identification of functional long non-coding RNAs in C. elegans. BMC Biol. 2019, 17, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Dai, L.; Ai, J.; Wang, Y.; Ren, F. Identification and functional prediction of cold-related long non-coding RNA (lncRNA) in grapevine. Sci. Rep. 2019, 9, 6638. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, X.; Han, K.; Zhang, G.; Wang, J.; Xie, K.; Xue, Q.; Fan, X. Analysis of long non-coding RNA and mRNA using RNA sequencing during the differentiation of intramuscular preadipocytes in chicken. PLoS ONE 2017, 12, e0172389. [Google Scholar]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Ma, T.; Chen, M.; Zou, Z.; Zhang, Z. The pseudogene-derived long non-coding RNA SFTA1P suppresses cell proliferation, migration, and invasion in gastric cancer. Biosci. Rep. 2018, 38, BSR20171193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milligan, M.J.; Lipovich, L. Pseudogene-derived lncRNAs: Emerging regulators of gene expression. Front. Genet. 2015, 5, 476. [Google Scholar] [CrossRef] [Green Version]
- Brenndörfer, M.; Boshart, M. Selection of reference genes for mRNA quantification in Trypanosoma brucei. Mol. Biochem. Parasitol. 2010, 172, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C. Regulation of gene expression in trypanosomatids: Living with polycistronic transcription. Open Biol. 2019, 9, 190072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mair, G.; Shi, H.; Li, H.; Djikeng, A.; Aviles, H.O.; Bishop, J.R.; Falcone, F.H.; Gavrilescu, C.; Montgomery, J.L.; Santori, M.I.; et al. A new twist in trypanosome RNA metabolism: Cis-splicing of pre-mRNA. RNA 2000, 6, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Chikne, V.; Gupta, S.K.; Doniger, T.; Cohen-Chalamish, S.; Waldman Ben-Asher, H.; Kolet, L.; Yahia, N.H.; Unger, R.; Ullu, E.; Kolev, N.G.; et al. The Canonical Poly (A) Polymerase PAP1 Polyadenylates Non-Coding RNAs and Is Essential for snoRNA Biogenesis in Trypanosoma brucei. J. Mol. Biol. 2017, 429, 3301–3318. [Google Scholar] [CrossRef]
- Siegel, T.N.; Gunasekera, K.; Cross, G.A.M.; Ochsenreiter, T. Gene expression in Trypanosoma brucei: Lessons from high-throughput RNA sequencing. Trends Parasitol. 2011, 27, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, A.; Caracausi, M.; Ricci, M.; Strippoli, P.; Vitale, L.; Pelleri, M.C. Identification of minimal eukaryotic introns through GeneBase, a user-friendly tool for parsing the NCBI Gene databank. DNA Res. 2015, 22, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederichs, S. The four dimensions of non-coding RNA conservation. Trends Genet. 2014, 30, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Karner, H.; Webb, C.-H.; Carmona, S.; Liu, Y.; Lin, B.; Erhard, M.; Chan, D.; Baldi, P.; Spitale, R.C.; Sun, S. Functional Conservation of LncRNA JPX Despite Sequence and Structural Divergence. J. Mol. Biol. 2020, 432, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Stijlemans, B.; Caljon, G.; Van Den Abbeele, J.; Van Ginderachter, J.A.; Magez, S.; De Trez, C. Immune Evasion Strategies of Trypanosoma brucei within the Mammalian Host: Progression to Pathogenicity. Front. Immunol. 2016, 7, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslett, M.; Aurrecoechea, C.; Berriman, M.; Brestelli, J.; Brunk, B.P.; Carrington, M.; Depledge, D.P.; Fischer, S.; Gajria, B.; Gao, X.; et al. TriTrypDB: A functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010, 38, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 January 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Lim, Y.W.; Edwards, R. Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 2012, 28, 433–435. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H.; Hester, J. Readr: Read Rectangular Text Data; Version 2.0.2; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Soneson, C.; Love, M.; Robinson, M. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2018, 35, 2084–2092. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.C.; Jolly, E.R. LncRNAs Are Differentially Expressed between Wildtype and Cell Line Strains of African Trypanosomes. Non-Coding RNA 2022, 8, 7. https://doi.org/10.3390/ncrna8010007
Kim HC, Jolly ER. LncRNAs Are Differentially Expressed between Wildtype and Cell Line Strains of African Trypanosomes. Non-Coding RNA. 2022; 8(1):7. https://doi.org/10.3390/ncrna8010007
Chicago/Turabian StyleKim, Hyung Chul, and Emmitt R. Jolly. 2022. "LncRNAs Are Differentially Expressed between Wildtype and Cell Line Strains of African Trypanosomes" Non-Coding RNA 8, no. 1: 7. https://doi.org/10.3390/ncrna8010007
APA StyleKim, H. C., & Jolly, E. R. (2022). LncRNAs Are Differentially Expressed between Wildtype and Cell Line Strains of African Trypanosomes. Non-Coding RNA, 8(1), 7. https://doi.org/10.3390/ncrna8010007