Convenient Preparation of Graphene Oxide from Expandable Graphite and Its Characterization by Positron Annihilation Lifetime Spectroscopy

 ,
,  ,
,  , and
, and
Abstract
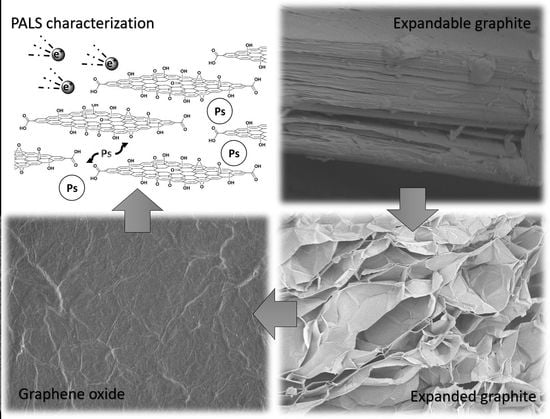
:
1. Introduction
2. Materials and Methods
Preparation of Graphene Oxide (GO)
3. Results and Discussion
3.1. Graphene Oxide from Expanded Graphite by a Simplified Hummers’ Method
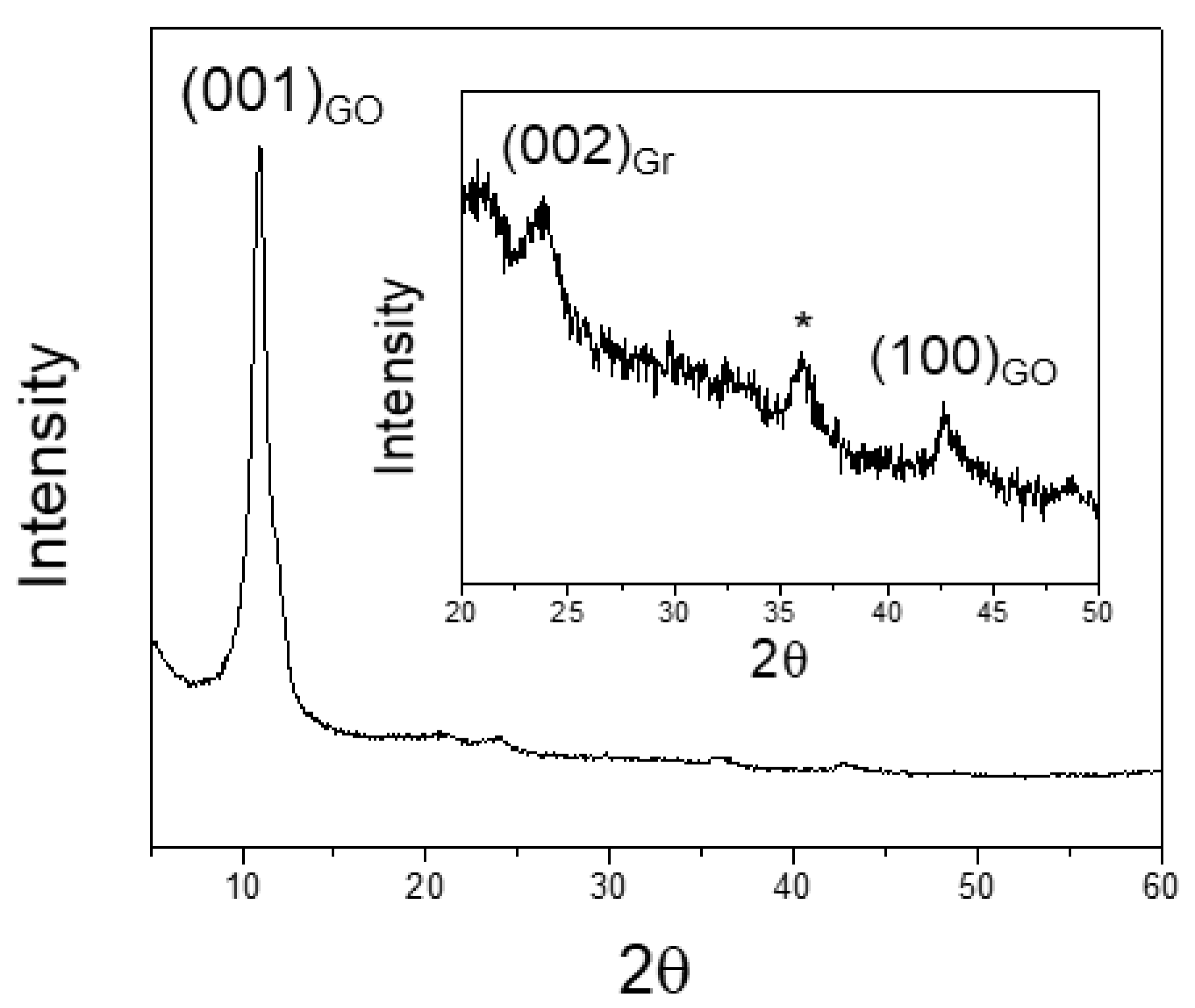
3.2. X-ray Powder Diffraction (XRPD) Results
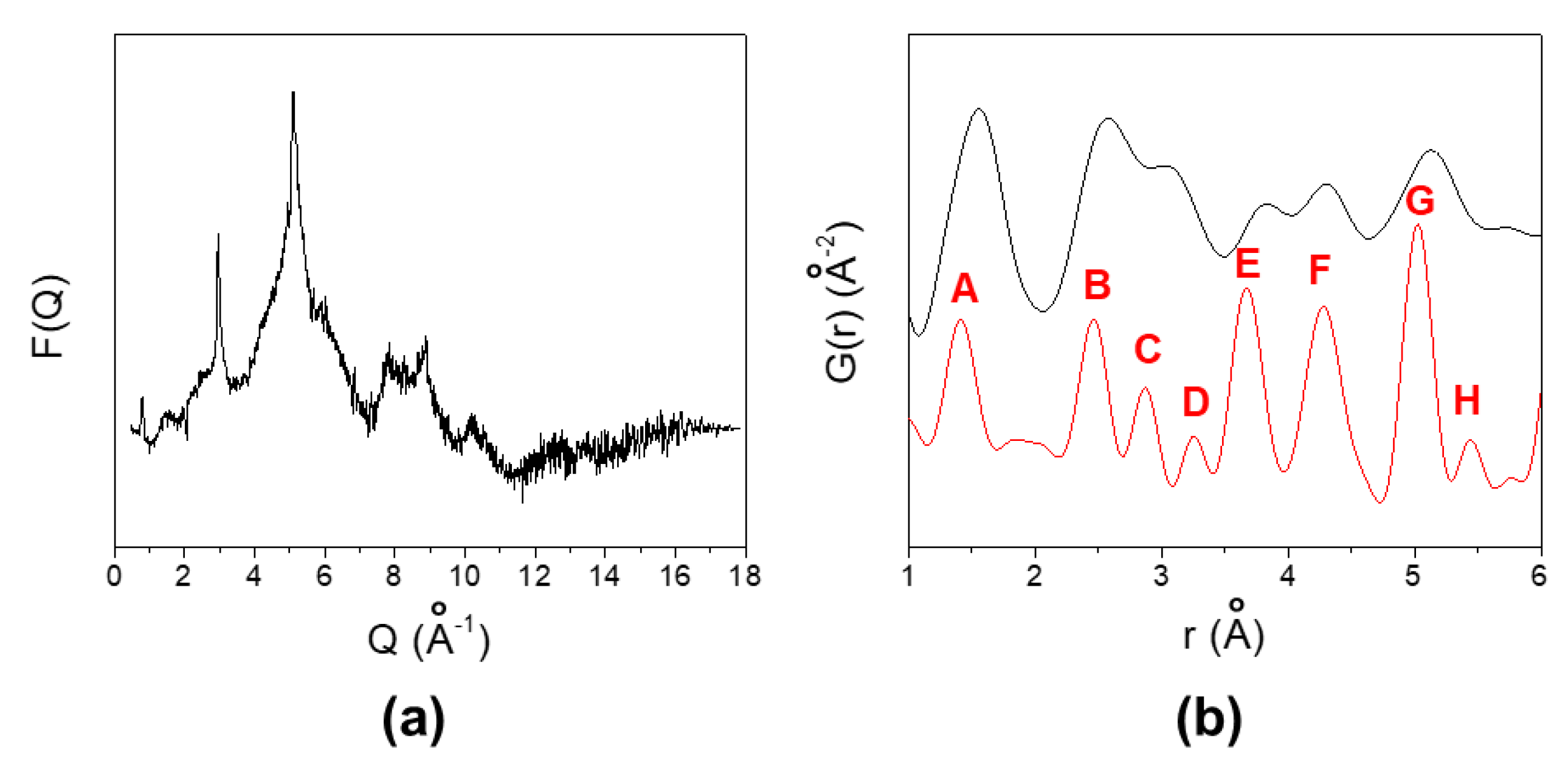
3.3. Positron Annihilation Lifetime Spectroscopy (PALS) of GO
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Tung, V.C.; Kaner, R.B. Honeycomb carbon: A review of graphene. Chem. Rev. 2010, 110, 132–145. [Google Scholar] [CrossRef]
- Georgakilas, V.; Otyepka, M.; Bourlinos, A.B.; Chandra, V.; Kim, N.; Kemp, K.C.; Hobza, P.; Zboril, R.; Kim, K.S. Functionalization of graphene: Covalent and non-covalent approaches, derivatives and applications. Chem. Rev. 2012, 112, 6156–6214. [Google Scholar] [CrossRef] [PubMed]
- Hummers, W.S.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Roisnel, T.; Rodríquez-Carvajal, J. WinPLOTR: A Windows Tool for Powder Diffraction Pattern Analysis. Mater. Sci. Forum 2001, 378–381, 118–123. [Google Scholar] [CrossRef]
- Ashiotis, G.; Deschildre, A.; Nawaz, Z.; Wright, J.P.; Karkoulis, D.; Picca, F.E.; Kieffer, J. The fast azimuthal integration Python library: PyFAI. J. Appl. Crystallogr. 2015, 48, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.B.; Sørensen, H.O.; Wright, J.P.; Goret, G.; Kieffer, J. FabIO: Easy access to two-dimensional X-ray detector images in Python. J. Appl. Crystallogr. 2013, 46, 537–539. [Google Scholar] [CrossRef]
- Egami, T.; Billinge, S.J.L. Underneath Bragg Peaks; Pergamon: New York, NY, USA, 2003. [Google Scholar]
- Qiu, X.; Thompson, J.W.; Billinge, S.J.L. PDFgetX2: A GUI-driven program to obtain the pair distribution function from X-ray powder diffraction data. J. Appl. Crystallogr. 2004, 37, 678. [Google Scholar] [CrossRef]
- Kansy, J. Microcomputer program for analysis of positron annihilation lifetime spectra. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 1996, 374, 235–244. [Google Scholar] [CrossRef]
- Focke, W.W.; Badenhorst, H.; Mhike, W.; Kruger, H.J.; Lombaard, D. Characterization of commercial expandable graphite fire retardants. Thermochim. Acta 2014, 584, 8–16. [Google Scholar] [CrossRef]
- Modesti, M.; Lorenzetti, A.; Simioni, F.; Camino, G. Expandable graphite as an intumescent flame retardant in polyisocyanurate-polyurethane foams. Polym. Degrad. Stab. 2002, 77, 195–202. [Google Scholar] [CrossRef]
- Sun, L.; Fugetsu, B. Mass production of graphene oxide from expanded graphite. Mater. Lett. 2013, 109, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Konkena, B.; Vasudevan, S. Understanding aqueous dispersibility of graphene oxide and reduced graphene oxide through pKa measurements. J. Phys. Chem. Lett. 2012, 3, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Arrais, A.; Diana, E.; Boccaleri, E. A study on the carbon soot derived from the wood combustion and on the relative alkali-extractable fraction. J. Mater. Sci. 2006, 41, 6035–6045. [Google Scholar] [CrossRef]
- Arrais, A.; Diana, E.; Rossetti, R.; Boccaleri, E. Spectral and chemical evidence for the direct formation of carboxylic groups in aerobically oxidised water-soluble fullerenes. Carbon 2007, 13, 2502–2510. [Google Scholar] [CrossRef]
- Hontoria-Lucas, C.; López-Peinado, A.J.; de López-González, J.D.; Rojas-Cervantes, M.L.; Martín-Aranda, R.M. Study of oxygen-containing groups in a series of graphite oxides: Physical and chemical characterization. Carbon N. Y. 1995, 33, 1585–1592. [Google Scholar] [CrossRef]
- Wu, J.B.; Lin, M.L.; Cong, X.; Liu, H.N.; Tan, P.H. Raman spectroscopy of graphene-based materials and its applications in related devices. Chem. Soc. Rev. 2018, 47, 1822–1873. [Google Scholar] [CrossRef] [Green Version]
- Kudin, K.N.; Ozbas, B.; Schniepp, H.C.; Prud’homme, R.K.; Aksay, I.A.; Car, R. Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett. 2008, 8, 36–41. [Google Scholar] [CrossRef]
- Krishnamoorthy, K.; Veerapandian, M.; Yun, K.; Kim, S.J. The chemical and structural analysis of graphene oxide with different degrees of oxidation. Carbon N. Y. 2013, 53, 38–49. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. Crystal Structure; Interscience Publishers: New York, NY, USA, 1963. [Google Scholar]
- Longhi, M.; Marzorati, S.; Checchia, S.; Sacchi, B.; Santo, N.; Zaffino, C.; Scavini, M. Sugar-based catalysts for oxygen reduction reaction. Effects of the functionalization of the nitrogen precursors on the electrocatalytic activity. Electrochim. Acta 2016, 222, 781–792. [Google Scholar] [CrossRef]
- Sormann, H.; Kontrym-Sznajd, G. Many-body effects on the electron-positron momentum density in simple and transition metals: Comparison with positron annihilation spectroscopy data. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 73, 075111. [Google Scholar] [CrossRef]
- Tuomisto, F.; Makkonen, I. Defect identification in semiconductors with positron annihilation: Experiment and theory. Rev. Mod. Phys. 2013, 85, 1583–1631. [Google Scholar] [CrossRef] [Green Version]
- Jean, Y.C.; Van Horn, J.D.; Hung, W.S.; Lee, K.R. Perspective of positron annihilation spectroscopy in polymers. Macromolecules 2013, 46, 7133–7145. [Google Scholar] [CrossRef]
- Panzarasa, G.; Aghion, S.; Soliveri, G.; Consolati, G.; Ferragut, R. Positron annihilation spectroscopy: A new frontier for understanding nanoparticle-loaded polymer brushes. Nanotechnology 2016, 27, 02LT03. [Google Scholar] [CrossRef] [PubMed]
- Panzarasa, G.; Aghion, S.; Marra, G.; Wagner, A.; Liedke, M.O.; Elsayed, M.; Krause-Rehberg, R.; Ferragut, R.; Consolati, G. Probing the Impact of the Initiator Layer on Grafted-from Polymer Brushes: A Positron Annihilation Spectroscopy Study. Macromolecules 2017, 50, 5574–5581. [Google Scholar] [CrossRef]
- Pandini, S.; Bignotti, F.; Baldi, F.; Sartore, L.; Consolati, G.; Panzarasa, G. Thermomechanical and large deformation behaviors of antiplasticized epoxy resins: Effect of material formulation and network architecture. Polym. Eng. Sci. 2017, 57, 553–565. [Google Scholar] [CrossRef]
- Dehghani, E.S.; Aghion, S.; Anwand, W.; Consolati, G.; Ferragut, R.; Panzarasa, G. Investigating the structure of crosslinked polymer brushes (brush-gels) by means of Positron Annihilation Spectroscopy. Eur. Polym. J. 2018, 99, 415–421. [Google Scholar] [CrossRef]
- Consolati, G.; Panzarasa, G.; Quasso, F. Morphology of free volume holes in an amorphous polyether-polyester polyurethane of biomedical interest. Polym. Test. 2018, 68, 208–212. [Google Scholar] [CrossRef]
- Tang, Y.P.; Paul, D.R.; Chung, T.S. Free-standing graphene oxide thin films assembled by a pressurized ultrafiltration method for dehydration of ethanol. J. Memb. Sci. 2014, 458, 199–208. [Google Scholar] [CrossRef]
- Gong, W.; He, D.; Tao, J.; Zhao, P.; Kong, L.; Luo, Y.; Peng, Z.; Wang, H. Formation of defects in the graphite oxidization process: A positron study. RSC Adv. 2015, 5, 88908–88914. [Google Scholar] [CrossRef]
- Tao, S.J. Positronium annihilation in molecular substances. J. Chem. Phys. 1972, 56, 5499. [Google Scholar] [CrossRef]
- Eldrup, M.; Lightbody, D.; Sherwood, J.N. The temperature dependence of positron lifetimes in solid pivalic acid. Chem. Phys. 1981, 63, 51–58. [Google Scholar] [CrossRef]
- Nakanishi, H.; Wang, S.J.; Jean, Y.C. Positron Annihilation Studies of Fluids; Sharma, S.C., Ed.; World Scientific: Singapore, 1988. [Google Scholar]
- Jasinska, B.; Koziol, A.E.; Goworek, T. Void shapes and o-Ps lifetimes in molecular crystals. Acta Phys. Pol. A 1999, 95, 557–561. [Google Scholar] [CrossRef]
- He, H.; Riedl, T.; Lerf, A.; Klinowski, J. Solid-state NMR studies of the structure of graphite oxide. J. Phys. Chem. 1996, 100, 19954–19958. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lifetime Components τ (ns) | Intensity Components I (%) |
|---|---|
| τ1 = 0.20 ± 0.02 | I1 = 54.2 ± 7.0 |
| τ2 = 0.44 ± 0.03 | I2 = 42.0 ± 5.0 |
| τ3 = 2.42 ± 0.04 | I3 = 3.8 ± 0.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panzarasa, G.; Consolati, G.; Scavini, M.; Longhi, M.; Quasso, F. Convenient Preparation of Graphene Oxide from Expandable Graphite and Its Characterization by Positron Annihilation Lifetime Spectroscopy. C 2019, 5, 6. https://doi.org/10.3390/c5010006
Panzarasa G, Consolati G, Scavini M, Longhi M, Quasso F. Convenient Preparation of Graphene Oxide from Expandable Graphite and Its Characterization by Positron Annihilation Lifetime Spectroscopy. C. 2019; 5(1):6. https://doi.org/10.3390/c5010006
Chicago/Turabian StylePanzarasa, Guido, Giovanni Consolati, Marco Scavini, Mariangela Longhi, and Fiorenza Quasso. 2019. "Convenient Preparation of Graphene Oxide from Expandable Graphite and Its Characterization by Positron Annihilation Lifetime Spectroscopy" C 5, no. 1: 6. https://doi.org/10.3390/c5010006
APA StylePanzarasa, G., Consolati, G., Scavini, M., Longhi, M., & Quasso, F. (2019). Convenient Preparation of Graphene Oxide from Expandable Graphite and Its Characterization by Positron Annihilation Lifetime Spectroscopy. C, 5(1), 6. https://doi.org/10.3390/c5010006