Carbon Materials as Cathode Constituents for Electrochemical CO2 Reduction—A Review


Abstract
:
1. Introduction
2. Carbon Black
2.1. Metal and Metal-Derived Particles Supported on Carbon Black
2.2. Molecular Catalysts
3. Mesoporous Carbon-Based Electrodes
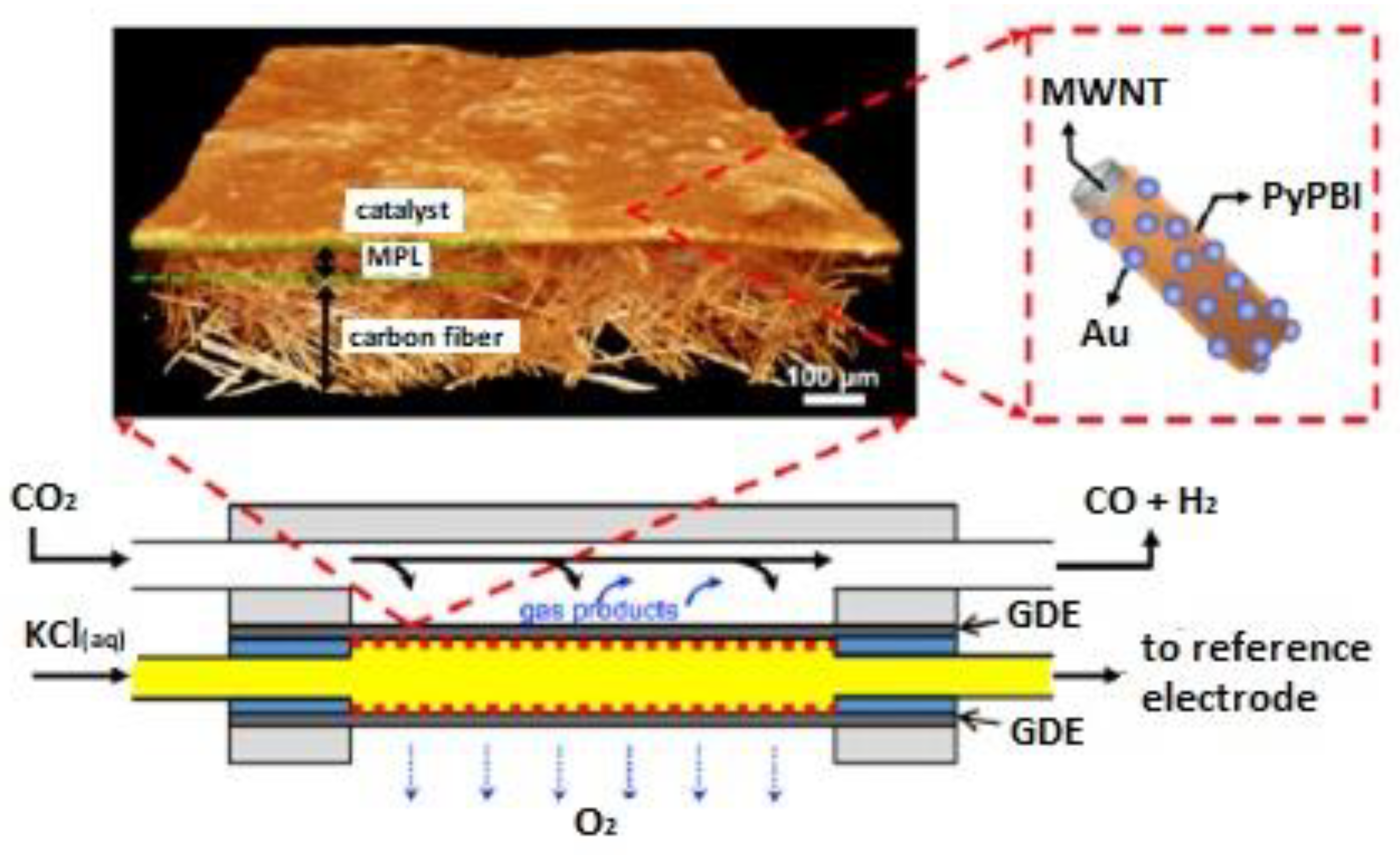
4. Carbon Fibers
5. Graphene
6. Graphene Derivatives
7. Carbon Nanotubes
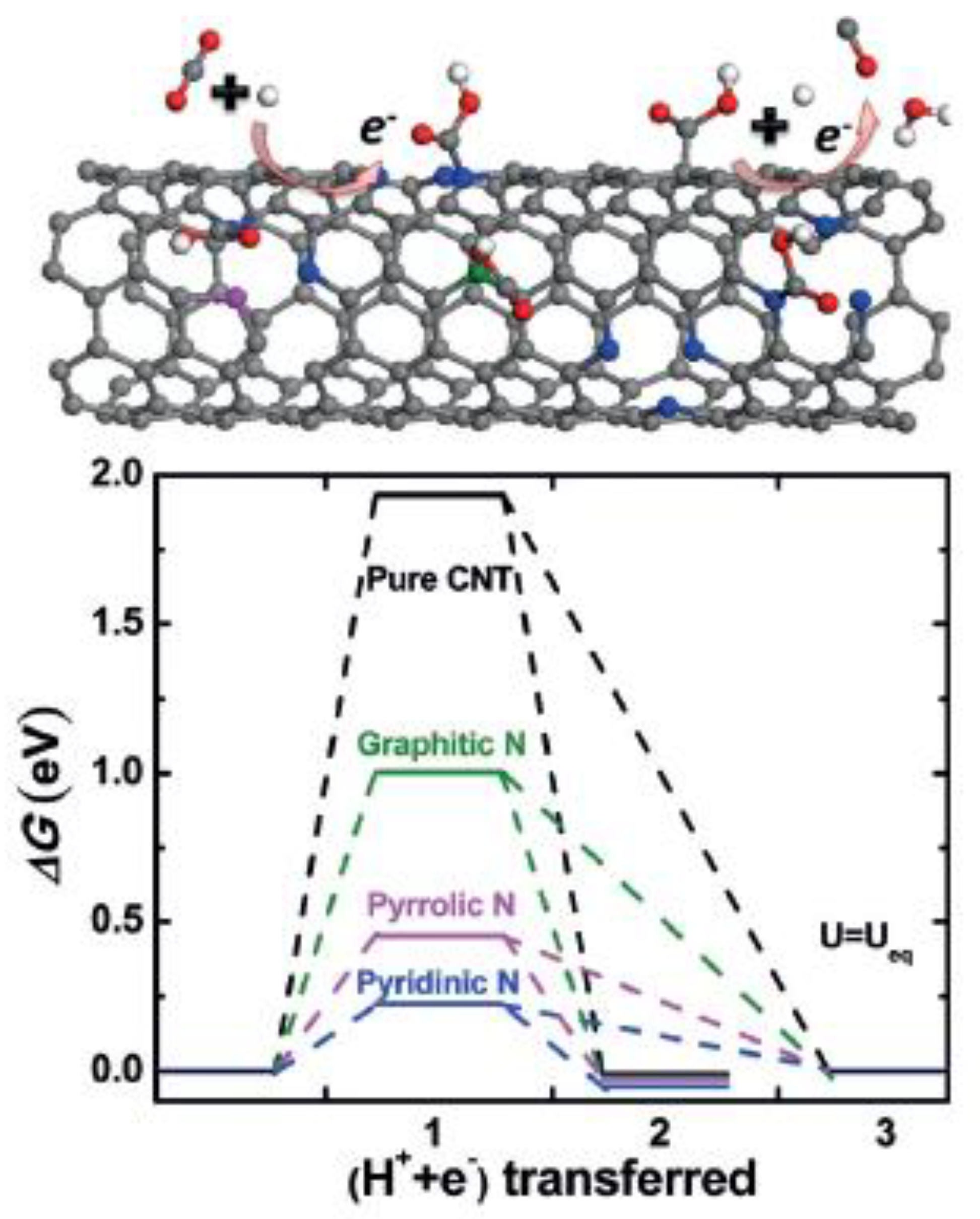
8. Nitrogen-Doped Carbon Nanotubes
9. Conclusions and Future Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Kätelhön, A.; Meys, R.; Deutz, S.; Suh, S.; Bardow, A. Climate change mitigation potential of carbon capture and utilization in the chemical industry. Proc. Natl. Acad. Sci. USA 2019, 116, 11187–11194. [Google Scholar] [CrossRef] [Green Version]
- Machado, A.S.R.; Nunes, A.V.M.; da Ponte, M.N. Carbon dioxide utilization—Electrochemical reduction to fuels and synthesis of polycarbonates. J. Supercrit. Fluids 2018, 134, 150–156. [Google Scholar] [CrossRef]
- Dean, J.A. Lange’s Handbook of Chemistry, 15th ed.; Mcgraw-Hill: New York, NY, USA, 1999; p. 1292. [Google Scholar]
- Mahmood, M.N.; Masheder, D.; Harty, C.J. Use of gas-diffusion electrodes for high-rate electrochemical reduction of carbon dioxide. I. Reduction at lead, indium- and tin-impregnated electrodes. J. Appl. Electrochem. 1987, 17, 1159–1170. [Google Scholar] [CrossRef]
- Hori, Y. Electrochemical CO2 reduction on metal electrodes. In Modern Aspects of Electrochemistry; White, R.E., Gamboa-Aldeco, M.E., Vayenas, C.G., Eds.; Springer: New York, NY, USA, 2008; Volume 42, pp. 89–189. [Google Scholar]
- Srinivasan, S. Experimental Methods in Low Temperature Fuel Cells. In Fuel Cells from Fundamentals to Applications; Springer: New York, NY, USA, 2006; pp. 268–306. [Google Scholar]
- Mot, B.; Hereijgers, J.; Duarte, M.; Breugelmans, T. Influence of flow and pressure distribution inside a gas diffusion electrode on the performance of a flow-by CO2 electrolyzer. Chem. Eng. J. 2019, 378. [Google Scholar] [CrossRef]
- Furuya, N.; Yamazaki, T.; Shibata, M. High performance Ru-Pd catalysts for CO2 reduction at gas diffusion electrodes. J. Electroanal. Chem. 1997, 43, 139–141. [Google Scholar] [CrossRef]
- Cook, R.L.; Macduff, R.C.; Sammells, A.F. High rate CO2 reduction to ethylene and methane using gas diffusion electrodes. J. Electrochem. Soc. 1990, 137, 607–608. [Google Scholar] [CrossRef]
- Shibata, M.; Yoshida, K.; Furuya, N. Elecfrochemical Synthesis of Urea at Gas-Diffusion Electrodes. J. Electrochem. Soc. 1998, 145, 595–600. [Google Scholar] [CrossRef]
- Hara, K.; Kudo, A.; Sakata, T.; Watanebe, M. High efficiency electrochemical reduction of carbon dioxide under high pressure on a gas diffusion electrode containing Pt catalysts. J. Electrochem. Soc. 1995, 142, L57–L59. [Google Scholar] [CrossRef]
- Hara, K.; Sakata, T. Electrocatalytic Formation of CH4 from CO2 on a Pt Gas Diffusion Electrode. J. Electrochem. Soc. 1997, 144, 539–545. [Google Scholar] [CrossRef]
- Hara, K.; Sakata, T. Large current density CO2 reduction under high pressure using gas diffusion electrodes. Bull. Chem. Soc. Jpn. 1997, 70, 571–576. [Google Scholar] [CrossRef]
- Zhao, G.; Jiang, T.; Han, B.; Li, Z.; Zhang, J.; Liu, Z.; He, J.; Wu, W. Electrochemical reduction of supercritical carbon dioxide in ionic liquid 1-n-butyl-3-methylimidazolium hexafluorophosphate. J. Supercrit. Fluids 2004, 32, 287–291. [Google Scholar] [CrossRef]
- Dufek, E.J.; Lister, T.E.; Stone, S.G.; McIlwain, M.E. Operation of a Pressurized System for Continuous Reduction of CO2. J. Electrochem. Soc. 2012, 159, F514–F517. [Google Scholar] [CrossRef]
- Manthiram, K.; Beberwyck, B.J.; Alivisatos, A.P. Enhanced Electrochemical Methanation of Carbon Dioxide with a Dispersible Nanoscale Copper Catalyst. J. Am. Chem. Soc. 2014, 136, 13319–13325. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Michalsky, R.; Metin, Ö.; Lv, H.F.; Guo, S.J.; Wright, C.J.; Sun, X.L.; Peterson, A.A.; Sun, S.H. Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, Y.J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A.A.; Sun, S. Active and selective conversion of CO2 to CO on ultrathin Au nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef]
- Douglas, R.; Kauffman, D.; Alfonso, C.; Matranga, H.F.; Qian, R.C.; Jin, R. Experimental and Computational Investigation of Au25 Clusters and CO2: A Unique Interaction and Enhanced Electrocatalytic Activity. J. Am. Chem. Soc. 2012, 134, 10237–10243. [Google Scholar]
- Liu, S.; Tao, H.; Zeng, L.; Liu, Q.; Xu, Z.; Liu, Q.; Luo, J.L. Shape-dependent electrocatalytic reduction of CO2 to CO on triangular silver nanoplates. J. Am. Chem. Soc. 2017, 139, 2160–2163. [Google Scholar] [CrossRef]
- Kim, C.; Jeon, H.S.; Eom, T.; Jee, M.S.; Kim, H.; Friend, C.M.; Min, B.K.; Hwanga, Y.J. Achieving selective and efficient electrocatalytic activity for CO2 reduction using immobilized silver nanoparticles. J. Am. Chem. Soc. 2015, 137, 13844–13850. [Google Scholar] [CrossRef]
- Li, Q.; Fu, J.; Zhu, W.; Chen, Z.; Shen, B.; Wu, L.; Xi, Z.; Wang, T.; Lu, G.; Zhu, J.J.; et al. Tuning Sn-catalysis for electrochemical reduction of CO2 to CO via the core/shell Cu/SnO2 structure. J. Am. Chem. Soc. 2017, 139, 4290–4293. [Google Scholar] [CrossRef]
- Azuma, M.; Hashimoto, K.; Hiramoto, M.; Watanabe, M.; Sakata, T. Electrochemical Reduction of Carbon Dioxide on Various Metal Electrodes in Low-Temperature Aqueous KHCO3 Media. J. Electrochem. Soc. 1990, 137, 1772–1778. [Google Scholar] [CrossRef]
- Noda, H.; Ikeda, S.; Oda, Y.; Imai, K.; Maeda, M.; Ito, K. Electrochemical reduction of carbon-dioxide at various metal-electrodes in aqueous potassium hydrogen carbonate solution. Bull. Chem. Soc. Jpn. 1990, 63, 2459–2462. [Google Scholar] [CrossRef] [Green Version]
- Ohkawa, K.; Hashimoto, K.; Fujishima, A.; Noguchi, Y.; Nakayama, S. Electrochemical reduction of carbon dioxide on hydrogen storing materials: Part 1. The effect of hydrogen absorption on the electrochemical behavior on palladium electrodes. Electroanal. Chem. 1993, 345, 445–456. [Google Scholar] [CrossRef]
- Hori, Y.; Wakebe, H.; Tsukamoto, T.; Koga, O. High Rate Gas Phase CO2. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim. Acta 1994, 39, 1833–1839. [Google Scholar] [CrossRef]
- Min, X.; Kanan, M.W. Pd-Catalyzed Electrohydrogenation of Carbon Dioxide to Formate: High Mass Activity at Low Overpotential and Identification of the Deactivation Pathway. J. Am. Chem. Soc. 2015, 137, 4701–4708. [Google Scholar] [CrossRef]
- Del Castillo, A.; Alvarez-Guerra, M.; Solla-Gullón, J.; Sáez, A.; Montiel, V.; Irabien, A. Sn nanoparticles on gas diffusion electrodes: Synthesis, characterization and use for continuous CO2 electroreduction to formate. J. CO2 Util. 2017, 18, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Wang, X.; Wang, Z.; Kang, P. Gas phase electrolysis of carbon dioxide to carbon monoxide using nickel nitride as the carbon enrichment catalyst. ACS Appl. Mater. Interfaces 2018, 10, 38024–38031. [Google Scholar] [CrossRef]
- Kutz, R.; Chen, Q.; Yang, H.; Sajjad, S.D.; Liu, Z.; Masel, R.I. Sustainion imidazolium-functionalized polymers for carbon dioxide electrolysis. Energy Technol. 2017, 5, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yang, H.; Kutz, R.; Masel, R.I. CO2 Electrolysis to CO and O2 at High Selectivity, Stability and Efficiency Using Sustainion Membranes. J. Electrochem. Soc. 2018, 165, J3371–J3377. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Savinova, E.R.; Yashnik, S.A.; Savinov, E.N.; Parmon, V.N. Gas-phase electrocatalytic reduction of CO2 to CO on carbon gas-diffusion electrode promoted by cobalt phthalocyanine. React. Kinet. Catal. Lett. 1992, 46, 249–254. [Google Scholar] [CrossRef]
- Mahmood, M.N.; Masheder, D.; Harty, C.J. Use of gas-diffusion electrodes for high-rate electrochemical reduction of carbon dioxide. IL Reduction at metal phthalocyanine-impregnated electrodes. J. Appl. Electrochem. 1987, 17, 1223–1226. [Google Scholar] [CrossRef]
- Shibata, M.; Furuya, N. Electrochemical synthesis of urea at gas-diffusion electrodes: Part VI. Simultaneous reduction of carbon dioxide and nitrite ions with various metallophthalocyanine catalysts. J. Electroanal. Chem. 2001, 507, 177–184. [Google Scholar] [CrossRef]
- Shibata, M.; Furuya, N. Simultaneous reduction of carbon dioxide and nitrate ions at gas-diffusion electrodes with various metallophthalocyanine catalysts. Electrochim. Acta 2003, 48, 3953–3958. [Google Scholar] [CrossRef]
- Wang, M.; Torbensen, K.; Salvatore, D.; Ren, S.; Joulié, D.; Dumoulin, F.; Mendoza, D.; Lassalle-Kaiser, B.; Isci, U.; Berlinguette, C.P.; et al. CO2 electrochemical catalytic reduction with a highly active cobalt phthalocyanine. Nat. Commun. 2019, 10, 3602. [Google Scholar] [CrossRef]
- Sonoyama, N.; Kirii, M.; Sakata, T. Electrochemical reduction of CO2 at metal-porphyrin supported gas diffusion electrodes under high pressure CO2. Electrochem. Commun. 1999, 1, 213–216. [Google Scholar] [CrossRef]
- Castelo-Quibén, J.; Bailón-García, E.; Pérez-Fernández, F.J.; Carrasco-Marín, F.; Pérez-Cadenas, A.F. Mesoporous carbon nanospheres with improved conductivity for electro-catalytic reduction of O2 and CO2. Carbon 2019, 155, 88–99. [Google Scholar] [CrossRef]
- Li, W.; Chen, D.; Li, Z.; Shi, Y.; Wan, Y.; Huang, J.; Yang, J.; Zhao, V.; Jiang, Z. Nitrogen enriched mesoporous carbon spheres obtained by a facile method and its application for electrochemical capacitor. Electrochem. Commun. 2007, 9, 569–573. [Google Scholar] [CrossRef]
- Song, Y.; Chen, W.; Zhao, C.; Li, S.; Wei, W.; Sun, Y. Metal-Free Nitrogen-Doped Mesoporous Carbon for Electroreduction of CO2 to Ethanol. Angew. Chem. Int. Ed. 2017, 56, 10840–10844. [Google Scholar] [CrossRef]
- Kuang, M.; Guan, A.; Gu, Z.; Han, P.; Qian, L.; Zheng, G. Enhanced N-doping in mesoporous carbon for efficient electrocatalytic CO2 conversion. Nano Res. 2019, 12, 2324–2329. [Google Scholar] [CrossRef]
- Morlanés, N.; Takanabe, K.; Rodionov, V. Simultaneous reduction of CO2 and splitting of H2O by a single immobilized cobalt phthalocyanine electrocatalyst. ACS Catal. 2016, 6, 3092–3095. [Google Scholar] [CrossRef]
- Rotundo, L.; Filippi, J.; Gobetto, R.; Miller, H.; Rocca, R.; Nervi, C.; Vizza, F. Electrochemical CO2 reduction in water at carbon cloth electrodes functionalized with a fac-Mn (apbpy)(CO)3Br complex. Chem. Commun. 2019, 55, 775–777. [Google Scholar] [CrossRef]
- Coeuret, F.; Vilar, E.O.; Cavalcanti, E.B. Carbon fibre cloth as an electrode material: Electrical conductivity and mass transfer. J. Appl. Electrochem. 2002, 32, 1175–1182. [Google Scholar] [CrossRef]
- Saito, N.; Aoki, K.; Usui, Y.; Shimizu, M.; Hara, K.; Narita, N.; Ogihara, N.; Nakamura, K.; Ishigaki, N.; Kato, H.; et al. Application of carbon fibers to biomaterials: A new era of nano-level control of carbon fibers after 30-years of development. Chem. Soc. Rev. 2011, 40, 3824–3834. [Google Scholar] [CrossRef] [PubMed]
- Magdesieva, T.V.; Yamamoto, T.; Tryk, D.A.; Fujishima, A. Electrochemical Reduction of CO2 with Transition Metal Phthalocyanine and Porphyrin Complexes Supported on Activated Carbon Fibers. J. Electrochem. Soc. 2002, 146, D89–D95. [Google Scholar] [CrossRef]
- Kumar, B.; Asadi, M.; Pisasale, D.; Sinha-Ray, S.; Rosen, B.A.; Haasch, R.; Abiade, J.; Yarin, A.L.; Salehi-Khojin, A. Renewable and metal-free carbon nanofibre catalysts for carbon dioxide reduction. Nat. Commun. 2013, 4, 2819–2826. [Google Scholar] [CrossRef]
- Reis-Machado, A.S.; Nunes da Ponte, M. CO2 Capture and Electrochemical Conversion. Curr. Opin. Green Sustain. Chem. 2018, 11, 86–90. [Google Scholar] [CrossRef]
- Yang, H.P.; Zhang, H.W.; Wu, Y.; Fan, L.D.; Chai, X.Y.; Zhang, Q.L.; Liu, J.H.; He, C.X. A core-shell-structured silver nanowire/nitrogen-doped carbon catalyst for enhanced and multifunctional electrofixation of CO2. ChemSusChem 2018, 11, 3905–3910. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.S.V.; Grigorieva, V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Rogers, C.; Perkins, W.S.; Veber, G.; Williams, T.E.; Cloke, R.R.; Fischer, F.R. Synergistic enhancement of electrocatalytic CO2 reduction with gold nanoparticles embedded in functional graphene nanoribbon composite electrodes. J. Am. Chem. Soc. 2017, 139, 4052–4061. [Google Scholar] [CrossRef] [Green Version]
- Geioushy, R.A.; Khaled, M.M.; Hakeem, A.S.; Khalid Alhooshani, K.; Basheer, C. High efficiency graphene/Cu2O electrode for the electrochemical reduction of carbon dioxide to ethanol. J. Electroanal. Chem. 2017, 785, 138–143. [Google Scholar] [CrossRef]
- Sreekanth, N.; Nazrulla, M.A.; Vineesh, T.V.; Sailaja, K.; Phani, K.L. Metal-free boron-doped graphene for selective electroreduction of carbon dioxide to formic acid/formate. Chem. Commun. 2015, 51, 16061–16064. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.X.; Chen, Y.B.; Hou, X.L.; Ma, C.Y.; Tan, T.W. Nitrogen-doped graphenes as efficient electrocatalysts for the selective reduction of carbon dioxide to formate in aqueous solution. Green Chem. 2016, 18, 3250–3256. [Google Scholar] [CrossRef]
- Wu, J.J.; Liu, M.J.; Sharma, P.P.; Yadav, R.M.; Ma, L.L.; Yang, Y.C.; Zou, X.L.; Zhou, X.D.; Vajtai, R.; Yakobson, B.I.; et al. Incorporation of Nitrogen Defects for Efficient Reduction of CO2 via Two-Electron Pathway on Three-Dimensional Graphene Foam. Nano Lett. 2016, 16, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhu, W.; Fu, J.; Zhang, H.; Wu, G.; Sun, S. Controlled assembly of Cu nanoparticles on pyridinic-N rich graphene for electrochemicalreduction of CO2 to ethylene. Nano Energy 2016, 24, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Yu, X.M.; Yuan, D.; Kuang, M.; Wang, Y.F.; Al-Enizi, A.M.; Zheng, G.F. Defective graphene for electrocatalytic CO2 reduction. J. Colloid Interface Sci. 2019, 534, 332–337. [Google Scholar] [CrossRef]
- Chai, G.L.; Guo, Z.X. Highly effective sites and selectivity of nitrogen doped graphene/CNT catalysts for CO2 electrochemical reduction. Chem. Sci. 2016, 7, 1268–1275. [Google Scholar] [CrossRef] [Green Version]
- Reske, R.; Mistry, H.; Behafarid, F.; Cuenya, B.R.; Strasser, P. Particle Size Effects in the Catalytic Electroreduction of CO2 on Cu Nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef]
- Mistry, H.; Reske, R.; Zeng, Z.H.; Zhao, J.; Greeley, J.; Strasser, P.; Cuenya, B.R. Exceptional Size-Dependent Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Liu, Y.; MacFarlane, D.R.; Wallace, G.G. Engineering Surface Amine Modifiers of Ultrasmall Gold Nanoparticles Supported on Reduced Graphene Oxide for Improved Electrochemical CO2 Reduction. Adv. Energy Mater. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cui, F.; Ross, M.B.; Kim, D.; Sun, Y.; Yang, P. CO2 electroreduction to hydrocarbons on ultrathin 5-fold twinned copper nanowires. Nano Lett. 2017, 17, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Alinajafi, H.A.; Ensafi, A.A.; Rezaei, B. Reduction of carbon dioxide to methanol on the surface of adenine functionalized reduced graphene oxide at a low potential. Int. J. Hydrog. Energy 2018, 43, 23262–23274. [Google Scholar]
- Ning, H.; Mao, Q.; Wang, W.; Yang, Z.; Wang, X.; Zhao, Q.; Song, Y.; Wu, M. N-doped reduced graphene oxide supported Cu2O nanocubes as high active catalyst for CO2 electro-reduction to C2H4. J. Alloy. Compd. 2019, 785, 7–12. [Google Scholar] [CrossRef]
- Zhou, X.; Micheroni, D.; Lin, Z.; Poon, C.; Li, Z.; Lin, W. Graphene-immobilized fac-Re(bipy)(CO)3Cl for syngas generation from carbon dioxide. ACS Appl. Mater. Interfaces 2016, 8, 4192–4198. [Google Scholar] [CrossRef]
- Choi, J.; Wagner, P.; Jalili, R.; Kim, J.; MacFarlane, D.R.; Wallace, G.G.; Officer, D.L. A porphyrin/graphene framework: A highly efficient and robust electrocatalyst for carbon dioxide reduction. Adv. Energy Mater. 2018, 8. [Google Scholar] [CrossRef]
- Yuan, J.; Zhi, W.Y.; Liu, L.; Yang, M.P.; Wang, H.; Lu, J.X. Electrochemical reduction of CO2 at metal-free N-functionalized graphene oxide electrodes. Electrochim. Acta 2018, 282, 694–701. [Google Scholar] [CrossRef]
- Xu, Y.; Sheng, K.; Li, C.; Shi, G. Self-Assembled Graphene Hydrogel via a One-Step Hydrothermal Process. ACS Nano 2010, 4, 4324–4330. [Google Scholar] [CrossRef]
- Choi, J.; Kim, J.; Wagner, P.; Gambhir, S.; Jalili, R.; Byun, S.; Sepidar, S.; Lee, Y.M.; MacFarlane, D.R.; Wallace, G.G.; et al. Energy efficient electrochemical reduction of CO2 to CO using a three-dimensional porphyrin/ graphene hydrogel. Energy Environ. Sci. 2019, 12, 747–755. [Google Scholar] [CrossRef]
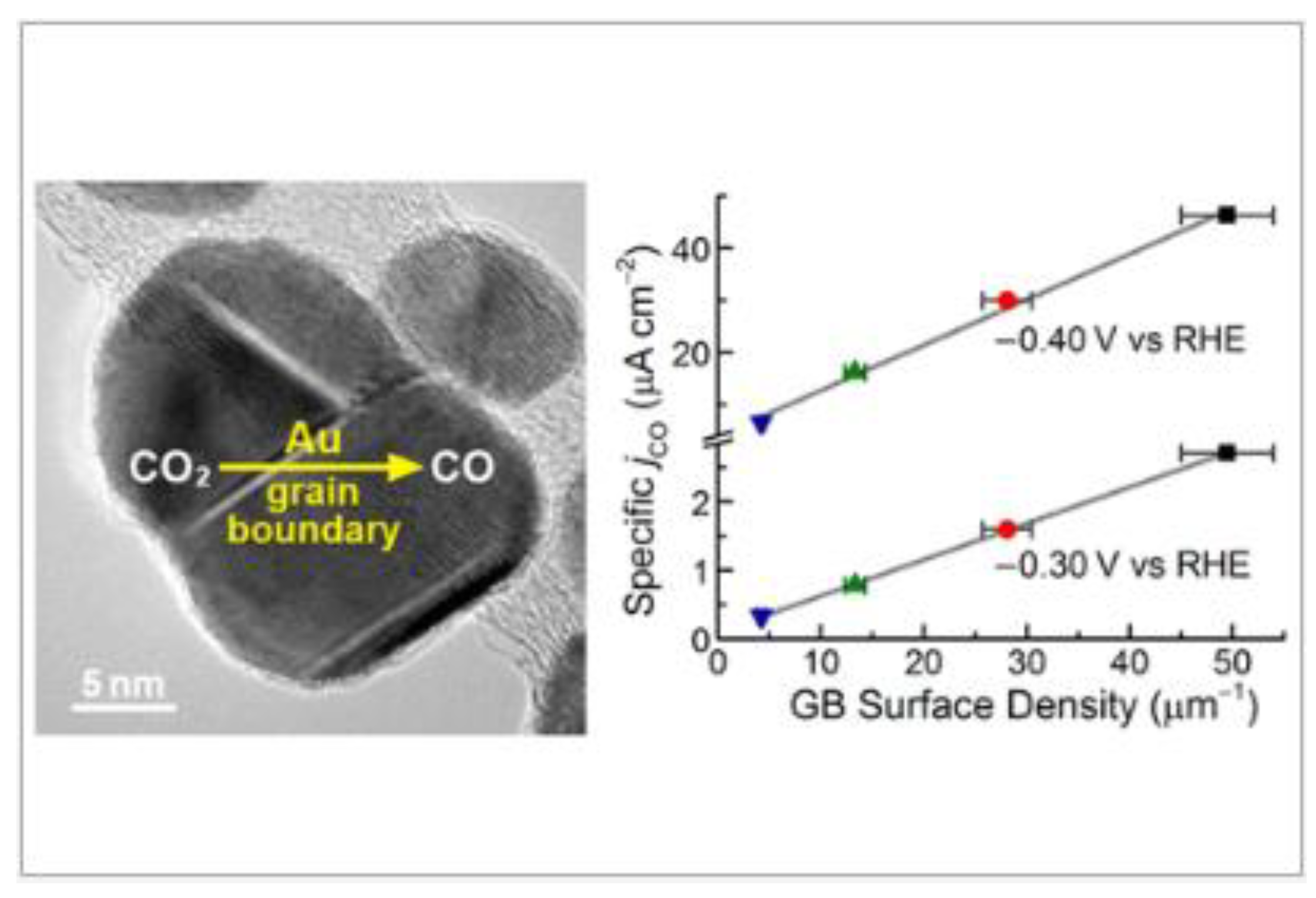
- Feng, X.; Kaili Jiang, K.; Fan, S.; Kanan, M.W. Grain-Boundary-Dependent CO2 Electroreduction Activity. J. Am. Chem. Soc. 2015, 137, 4606–4609. [Google Scholar] [CrossRef]
- Jhong, H.R.M.; Tornow, C.E.; Kim, C.; Verma, S.; Oberst, J.L.; Anderson, P.S.; Gewirth, A.A.; Fujigaya, T.; Nakashima, N.; Kenis, P.J.A. Gold Nanoparticles on Polymer-Wrapped Carbon Nanotubes: An Efficient and Selective Catalyst for the Electroreduction of CO2. ChemPhysChem 2017, 18, 3274–3279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Hamasaki, Y.; Kim, C.R.; Huang, W.; Lu, S.; Jhong, H.R.M.; Gewirth, A.A.; Fujigaya, T.; Nakashima, N.; Kenis, P.J.A. Insights into the low overpotential electroreduction of CO2 to CO on a supported gold catalyst in an alkaline flow electrolyzer. ACS Energy Lett. 2018, 3, 193–198. [Google Scholar] [CrossRef]
- Zhao, C.C.; Wang, J.L.; Goodenough, J.B. Comparison of electrocatalytic reduction of CO2 to HCOOH with different tin oxides on carbon nanotubes. Electrochem. Commun. 2016, 65, 9–13. [Google Scholar] [CrossRef]
- Ma, S.; Luo, R.; Gold, J.I.; Yu, A.Z.; Kim, B.; Kenis, P.J.A. Carbon nanotube containing Ag catalyst layers for efficient and selective reduction of carbon dioxide. J. Mater. Chem. A 2016, 4, 8573–8578. [Google Scholar] [CrossRef]
- Bashir, S.; Hossain, S.S.; Rahmane, S.; Ahmed, S.; Al-Ahmed, A.; Hossain, M.M. Electrocatalytic reduction of carbon dioxide on SnO2/MWCNT in aqueous electrolyte solution. J. CO2 Util. 2016, 16, 346–353. [Google Scholar] [CrossRef]
- Marepally, B.C.; Ampelli, C.; Genovese, C.; Tavella, F.; Veyre, L.; Quadrelli, E.A.; Perathoner, S.; Centi, G. Role of small Cu nanoparticles in the behaviour of nanocarbon-based electrodes for the electrocatalytic reduction of CO2. J. CO2 Util. 2017, 21, 534–542. [Google Scholar] [CrossRef]
- Walsh, J.J.; Neri, G.; Smith, C.L.; Cowan, A.J. Electrocatalytic CO2 reduction with a membrane supported manganese catalyst in aqueous solution. Chem. Commun. 2014, 50, 12698–12701. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.J.; Smith, C.L.; Neri, G.; Whitehead, G.F.S.; Robertson, C.M.; Cowan, A.J. Improving the efficiency of electrochemical CO2 reduction using immobilized manganese complexes. Faraday Discuss. 2015, 183, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Aoi, S.; Mase, K.; Ohkubo, K.; Fukuzumi, S. Selective electrochemical reduction of CO2 to CO with a cobalt chlorin complex adsorbed on multi-walled carbon nanotubes in water. Chem. Commun. 2015, 50, 10226–10228. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.M.; Ronne, M.H.; Pedersen, S.U.; Skrydstrup, T.; Daasbjerg, K. Enhanced Catalytic Activity of Cobalt Porphyrin in CO2. Electroreduction upon Immobilization on Carbon Materials. Angew. Chem. Int. Ed. 2017, 56, 6468–6472. [Google Scholar] [CrossRef]
- Zhang, X.; Zishan Wu, Z.; Zhang, X.; Li, L.; Yanyan Li, Y.; Xu, H.; Li, X.; Xiaolu Yu, X.; Zhang, Z.; Liang, Y.; et al. Highly selective and active CO2 reduction electrocatalysts based on cobalt phthalocyanine/carbon nanotube hybrid structures. Nat. Commun. 2017, 8, 14675–14682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, N.; Wang, Y.; Ma, L.; Wen, J.; Li, J.; Zheng, H.; Nie, K.; Wang, X.; Zhao, F.; Li, Y.; et al. Supported cobalt phthalocyanine for high-performance electrocatalytic CO2 reduction. Chem 2017, 3, 652–664. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Chen, L.; Lau, T.-C.; Robert, M. Hybrid Co quaterpyridine complex/carbon nanotube catalytic material for CO2 reduction in water. Angew. Chem. Int. Ed. 2018, 57, 7769–7773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kang, P.; Ubnoske, S.; Brennaman, M.K.; Song, N.; House, R.L.; Glass, J.T.; Meyer, J. Polyethylenimine-enhanced electrocatalytic reduction of CO2 to formate at nitrogen-doped carbon nanomaterials. J. Am. Chem. Soc. 2014, 136, 7845–7848. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jia, J.; Song, P.; Wang, Q.; Li, D.; Min, S.; Qian, C.; Wang, L.; Li, Y.F.; Ma, C.; et al. Efficient Electrocatalytic Reduction of CO2 by Nitrogen-Doped Nanoporous Carbon/Carbon Nanotube Membranes – A Step Towards the Electrochemical CO2 Refinery. Angew. Chem. Int. Ed. 2017, 56, 7847–7852. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.J.; Yadav, R.M.; Liu, M.J.; Sharma, P.P.; Tiwary, C.S.; Ma, L.L.; Zou, X.L.; Zhou, X.D.; Yakobson, B.I.; Lou, J.; et al. Achieving Highly Efficient, Selective, and Stable CO2 Reduction on Nitrogen-Doped Carbon Nanotubes. ACS Nano 2015, 9, 5364–5371. [Google Scholar] [CrossRef]
- Sumpter, B.G.; Huang, J.; Meunier, V.; Romo-Herrera, J.M.; Cruz-Silva, E.; Terrones, H.; Terrones, M. A theoretical and experimental study on manipulating the structure and propoerties of carbon nanotubes using substitutional dopants. Int. J. Quantum Chem. 2009, 109, 97–118. [Google Scholar] [CrossRef]
- Sharma, P.P.; Wu, J.J.; Yadav, R.M.; Liu, M.J.; Wright, C.J.; Tiwary, C.S.; Yakobson, B.I.; Lou, J.; Ajayan, P.M.; Zhou, X.D. Nitrogen-Doped Carbon Nanotube Arrays for High-Efficiency Electrochemical Reduction of CO2: On the Understanding of Defects, Defect Density, and Selectivity. Angew. Chem. Int. Ed. 2015, 54, 13701–13705. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Smith, S.C.; Du, A.J.; Zhu, Z.H. Electrocatalytically Switchable CO2 Capture: First Principle Computational Exploration of Carbon Nanotubes with Pyridinic Nitrogen. ChemSusChem 2014, 7, 435–441. [Google Scholar] [CrossRef]
- Cui, X.Q.; Pan, Z.Y.; Zhang, L.J.; Peng, H.S.; Zheng, G.F. Selective etching of nitrogen-doped carbon by steam for enhanced electrochemical CO2 reduction. Adv. Energy Mater. 2017, 7, 1701456. [Google Scholar] [CrossRef]
- Liu, K.H.; Zhong, H.X.; Yang, X.Y.; Bao, D.; Meng, F.L.; Yan, J.L.; Zhang, X.B. Composition-tunable synthesis of “clean” syngas via a one-step synthesis of metal-free pyridinic-N-enriched self-supported CNTs: The synergy of electrocatalyst pyrolysis temperature and potential. Green Chem. 2017, 19, 4284–4288. [Google Scholar] [CrossRef]
- Diaz, L.A.; Gao, N.; Adhikari, B.; Lister, T.E.; Dufek, E.J.; Wilson, A.D. Electrochemical production of syngas from CO2 captured in switchable polarity solvents. Green Chem. 2018, 20, 620–626. [Google Scholar] [CrossRef]
- Messias, S.; Sousa, M.M.; Nunes da Ponte, M.; Rangel, C.M.; Pardal, T.; Reis-Machado, A.S. Electrochemical production of syngas from CO2 at pressures up to 30 bar in electrolytes containing ionic liquid. React. Chem. Eng. 2019, 4, 1982–1990. [Google Scholar] [CrossRef] [Green Version]










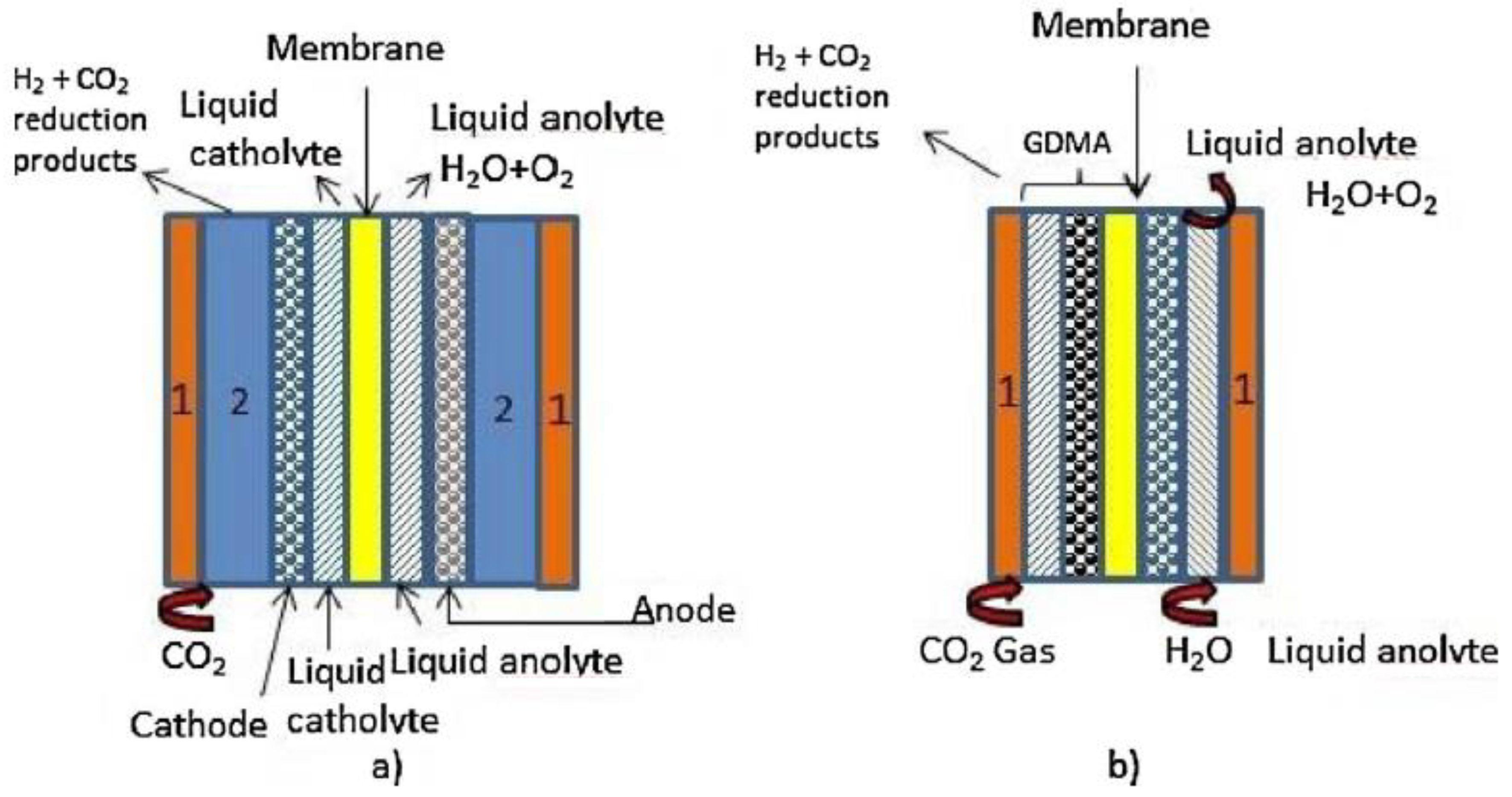
{kind=link}
{kind=link}
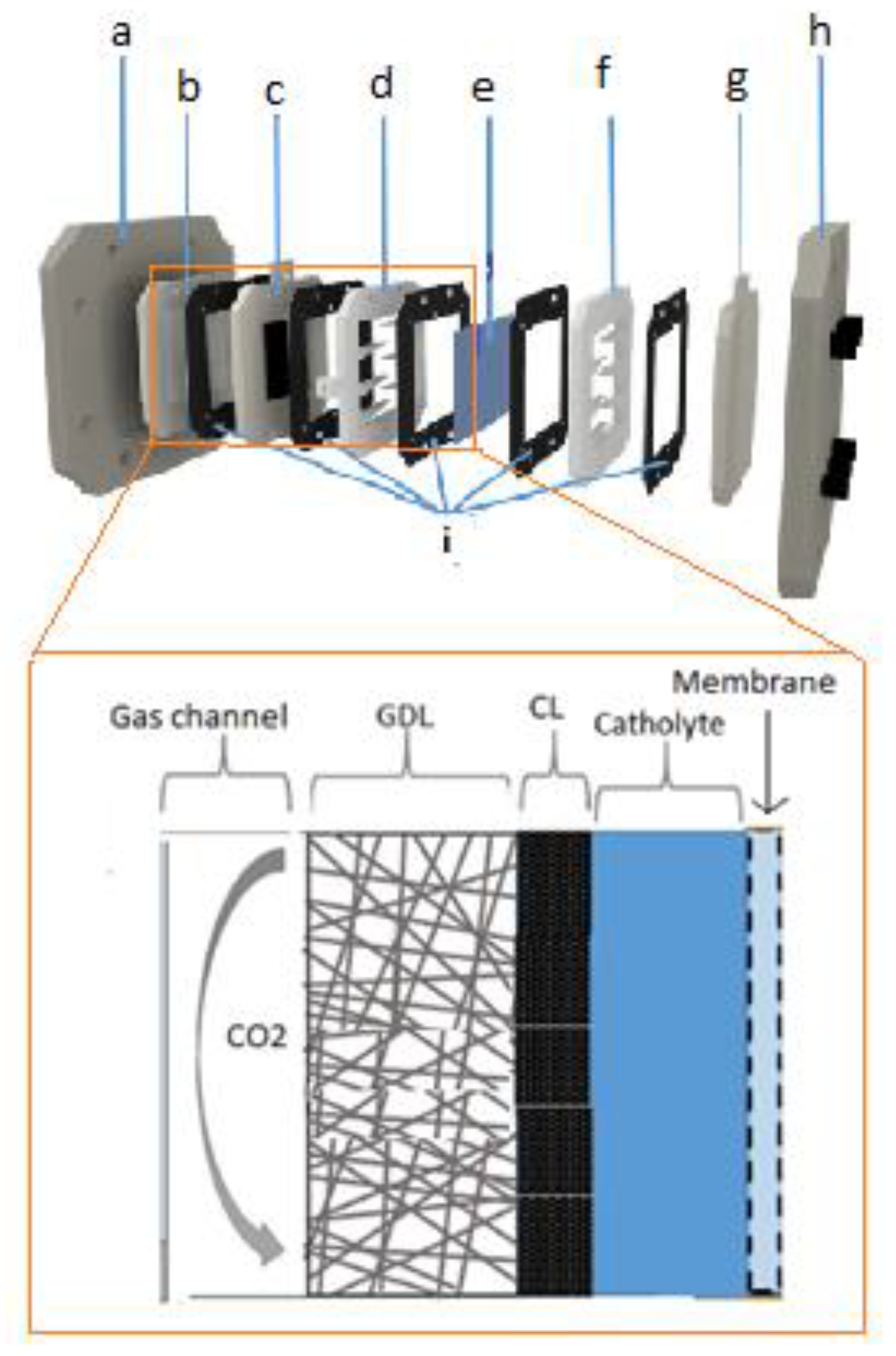{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of Carbon | Catalyst | Current Density (mA/cm2) | Potential/ Voltage | CO2RR Products | Faradaic Efficiency | Reference |
|---|---|---|---|---|---|---|
| Supported Metal and Metal Derived Particles | ||||||
| Carbon Black | Pb on Vulcan 72 | 115 | −2.7 to −3.0V vs. SCE | Formic acid | 97% | Mahmood et al. [6] |
| Cu | 667 | −4.75 V vs. Ag/AgCL | Ethene | 53% | Cook et al. [11] | |
| Cu | 300 | −1.25 V vs. SHE | Ethylene and ethanol | - | Ikeda et al. [7] | |
| Ru-Pd alloys | 80 | −1.1V vs. SHE | Formic acid | 90% | Furuya et al. [10] | |
| Pt | 600 | −1.93 V vs. Ag/AgCL | Methane | 38.8% | Hara et al. [13] | |
| Au, 8 nm NP | 3A/g | −0.67 V vs. RHE | Carbon monoxide | 90% | Zhu et al. [19] | |
| Au 500 nm nanowires | 8 | −0.35 V vs. RHE | Carbon monoxide | 94% | Zhu et al. [20] | |
| Au25 Clusters | - | −1 V vs. RHE | Carbon monoxide | 100% | Kauffman et al. [21] | |
| Triangular Ag nanoplates | 1.25 | −0.86 V vs. RHE | Carbon monoxide | 97% | Liu et al. [22] | |
| Cysteamine anchored 5 nmAg nanoparticles | 1 | −0.35 V vs. RHE | Carbon monoxide | ~80% | Kim et al. [23] | |
| Core/Shell Cu/SnO2 Structure | 4.6 | −0.7 V vs. RHE | Carbon monoxide | 93% | Li et al. [24] | |
| Pd 5 nm nanoparticles | 2.2 to 4.15 | >−0.2 V vs. RHE | Formic acid | >95% | Min et al. [29] | |
| Sn nanoparticles | 200 | −1.4 V vs. Ag/AgCl/4 V cell voltage | Formic acid | 54.1% | Del Castillo et al. [30] | |
| Ni nitride | 23.3 | −0.90 V vs. RHE | Carbon monoxide | 92.5% | Hou et al. [31] | |
| Ag 20 nm nanoparticles | 200 | 3 V cell voltage | Carbon monoxide | 98% | Liu et al. [33] | |
| Molecular Catalysts | ||||||
| Co-Phthalocyanine | 80 | −4.39 V vs. Ag/AgCl | Carbon monoxide | 100% | Savinova et al. [35] | |
| Co-Phtahlocyanine | 137 22 | −2.2 V vs. SCE −1.5 V vs. SCE | Carbon monoxide | 14% 100% | Mahmood et al. [36] | |
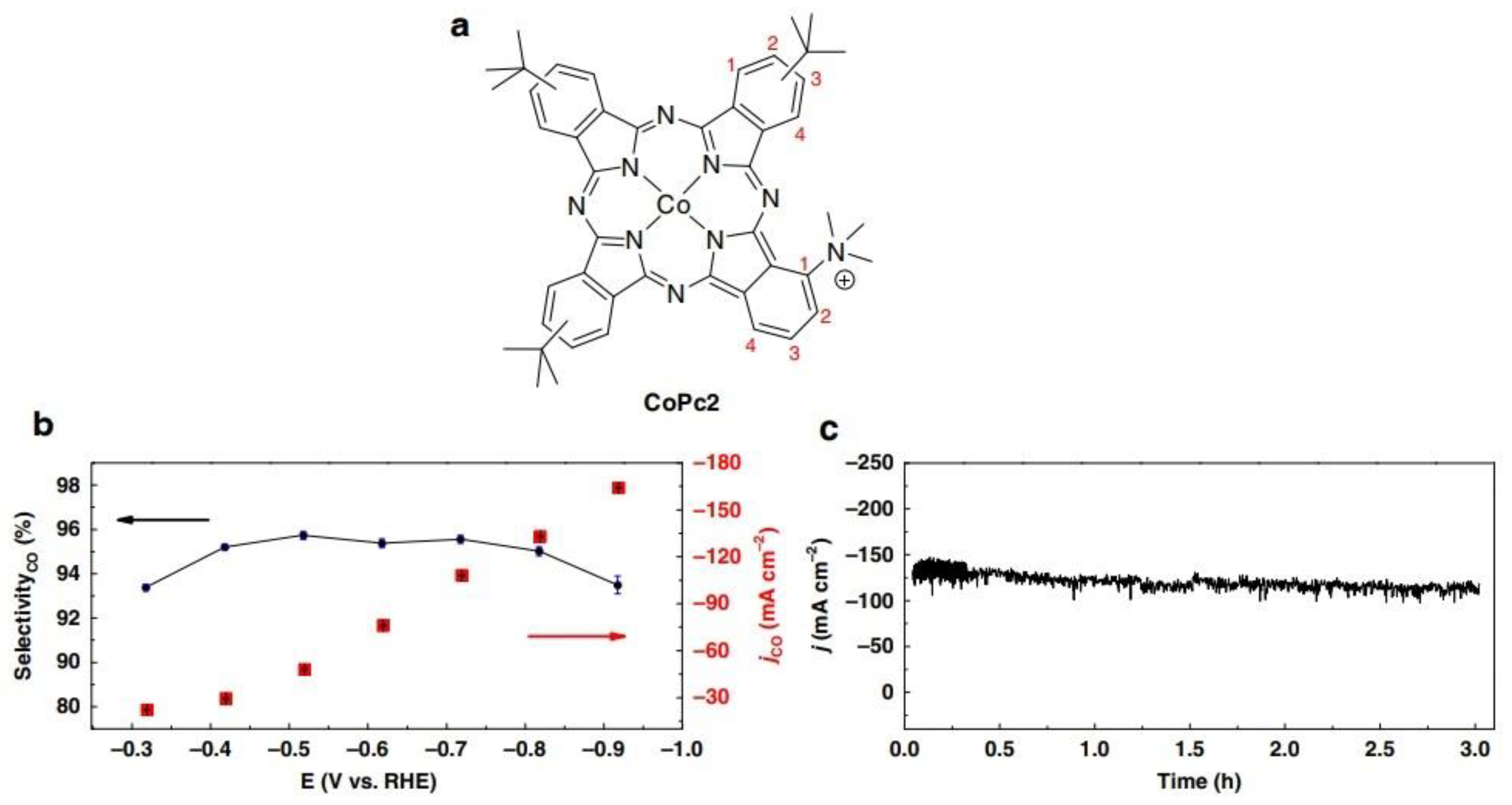
| Co Phthalocyanine2 | 165 | −0.92 V vs. RHE | Carbon monoxide | 95% | Wang et al. [39] | |
| Mesoporous Carbon | Carbon nanospheres impregnated with Ni particles | - | −1.6 V vs. Ag/AgCl | C1-C4 hydrocarbons | 0.3% | Castelo-Quibén et al. [41] |
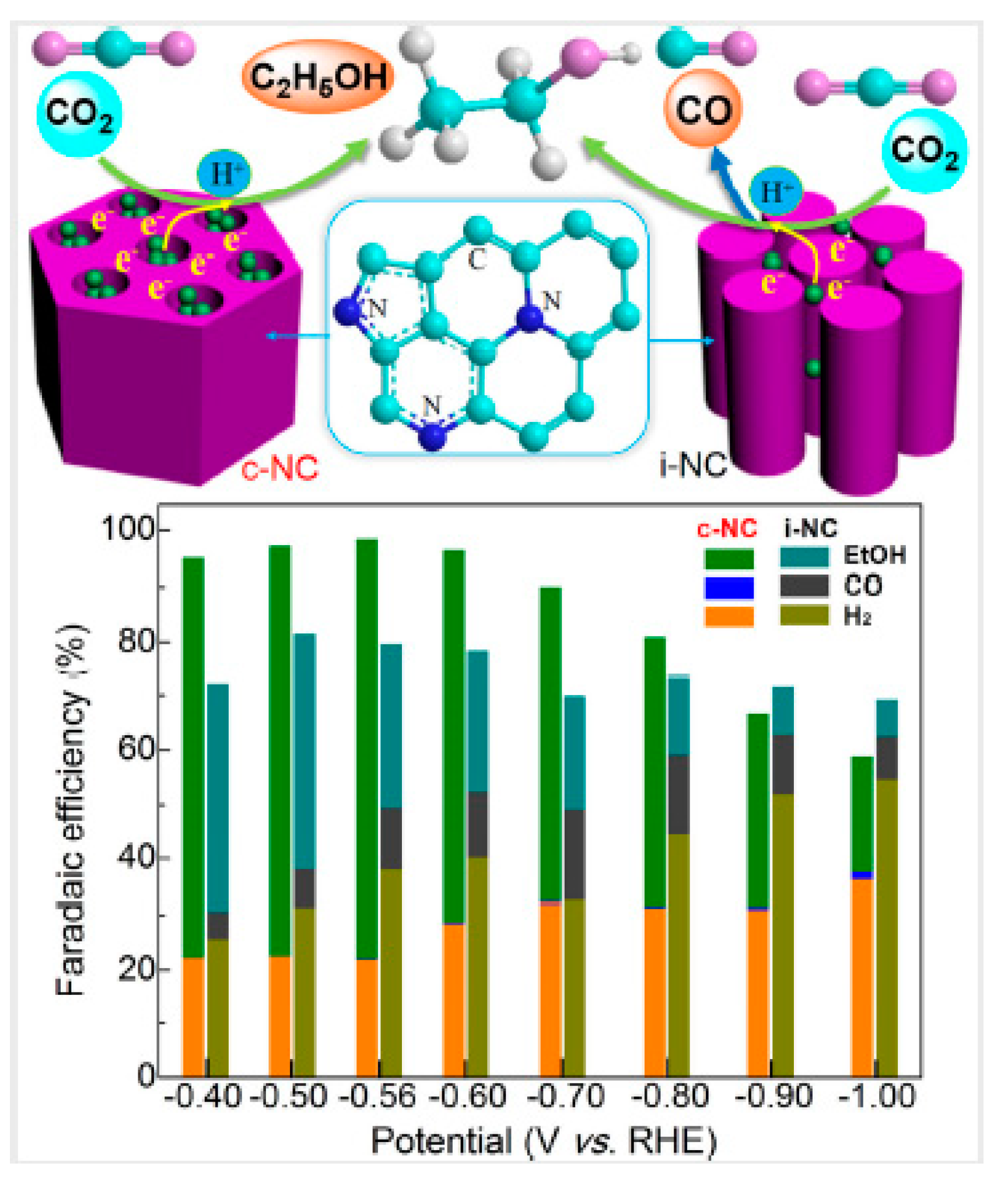
| Nitrogen doped mesoporous carbon | 0.25 | −0.56 V vs. RHE | Ethanol | 77% | Song et al. [43] | |
| mesoporous N-doped carbon | 6.8 | −0.58 V vs. RHE | Carbon monoxide | ~92% | Kuang et al. [44] | |
| Carbon Fibers | CoFPc immobilized on carbon cloth | 1–6 | 2–3 V cell voltage | Carbon monoxide | ~90% | Morlanés et al. [45] |
| fac-Mn(apbpy)(CO)3Br attached on carbon cloth | 0.9 | −1.35 V vs. Ag/AgCl | syngas | 60% for CO | Rotundo et al. [46] | |
| porphyrin and phthalocyanine | 70 | −1.5 V vs. SCE | Carbon monoxide | 70% | Magdesieva et al. [49] | |
| N-doped carbon nanofibers | 0.5 | −0.573 V vs. RHE | Carbon monoxide | 98% | Kumar et al. [50] | |
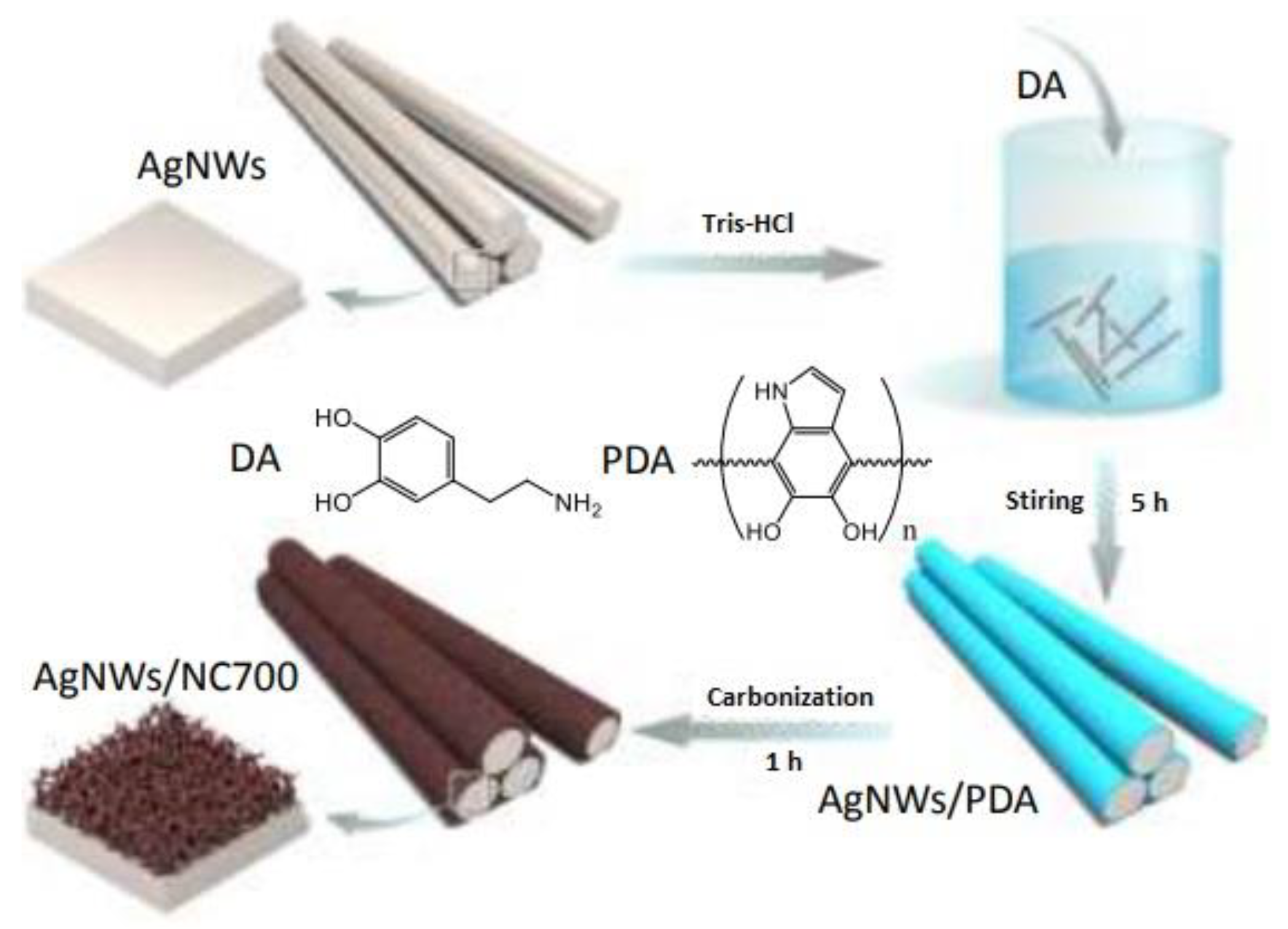
| N-doped carbon fibers with a core of Ag nanowires | - | −0.8 V vs. RHE | Carbon monoxide Hydrogen | 96% | Yang et al. [52] | |
| Au nanoparticles supported on graphene nanoribbon | ~4–6 | −0.47 V vs. RHE | Carbon monoxide | >60% | Rogers et al. [54] | |
| Graphene | Cu2O nanoparticles supported on graphene | - | −0.9 V vs. Ag/AgCl | Ethanol | 9.93% | Geioushy et al. [55] |
| N-doped graphene | 7.5 | −0.84 vs. Ag/AgCl | Formate | 70–63% | Wang et al. [57] | |
| N-doped 3D-graphene foam | 1.8 | −0.58 V vs. RHE | Carbon monoxide Formic acid | 70% 3.0% | Wu et al. [58] | |
| B-doped graphene | 0.5 | −1.4 V vs. SCE | Formate | 66% | Sreekanth et al. [56] | |
| N-doped graphene | - | −0.9 V vs. RHE | Formate | 65% | Li et al. [59] | |
| Cu nanoparticles dispersed on n-doped graphene | −0.9 V vs. RHE | Ethylene | 19% | Li et al. [59] | ||
| N-defective graphene | 1.3 | −0.6 V vs. RHE | Carbon monoxide | ~84% | Han et al. [60] | |
| Graphene Derivatives | Amine modified Au nanoparticles supported on rGO | 6 | −0.7 V vs. RHE | Carbon monoxide | >60% | Zhao et al. [64] |
| Cu nanowires wrapped in rGO | 8 | −1.25 V vs. RHE | Methane | 55% | Li et al. [65] | |
| Pt deposited on adenine-rGO | 0.5 | −0.3V vs. Ag/AgCl | Methanol | >85% | Hossein et al. [66] | |
| Cu2O/N-doped rGO | 12 | −1.4 V vs. RHE | Ethylene | 19.7% | Ning et al. [67] | |
| porphyrin/graphene framework | 1.68 | −0.54V vs. RHE | Carbon monoxide | 98.7% | Choi et al. [69] | |
| Vitamin B6 grafted on GO sheet | 0.25 | −0.4 V vs. RHE | Ethanol | 37% | Yuan et al. [70] | |
| iron porphyrin-based rGO hydrogel | 0.42 | −0.39 V vs. RHE | Carbon monoxide | 96% | Choi et al. [72] | |
| Au nanoparticles supported on MWCNT | 160 | −1.78 V vs. Ag/AgCl | Carbon monoxide | 60% | Jhong et al. [74] | |
| Au nanoparticles supported on MWCNT | 158 | −0.55 V vs. RHE | Carbon monoxide | 85% | Verma et al. [75] | |
| SnOx supported on MWCNT | 5–10 | −1.4 V vs. SCE | Formic acid | 60% | Zhao et al. [76] | |
| Carbon nanotubes | MWCNTs covered with a layer of Ag | 350 | −3 V cell voltage | Carbon monoxide | 95% | Ma et al. [77] |
| SnO2/MWCNT with 20%weight of SnO2 | 80 | −1.7 V vs. SCE | Formate | 27.2% | Bashir et al. [78] | |
| Immobilized Mn complex in Nafion/MWCNT | 2.65 | −15 V vs. SCE | Carbon monoxide | 24% | Walsh et al. [80] | |
| cobalt(II) chlorin complex | - | −1.1 V vs. NHE | Carbon monoxide | 89% | Aoi et al. [82] | |
| Co phthalocyanine funcionalised with CN groups anchored on CNT | 15 | −0.63 V vs. RHE −0.63 V vs. RHE | Carbon monoxide | >95% | Zhang et al. [84] | |
| CNT immobilized on cobalt polyphthalocyanine sheath | 18.7 | −0.5 V vs. RHE | Carbon monoxide | >80% | Han et al. [85] | |
| Co complex/CNT composite material | 9.3 | −0.48 V vs. RHE | Carbon monoxide | 100% | Wang et al. [86] | |
| CNT embedded in a carbon matrix | 6 | −0.8 V vs. RHE | Formate | 81% | Wang et al. [88] | |
| Nitrogen-doped carbon nanotubes | PEI-N-doped CNT | 7.2 | −1.8 V vs. SCE | Formate | 85% | Zhang et al. [87] |
| N-doped CNT | 1 | −0.8 V vs. RHE | Carbon monoxide | 80% | Wu et al. [89] | |
| N-doped CNT with pyridinic (1.1 at%) and graphitic (3.5 at%) | 2 | −1.05 V cell voltage (corrected for IR drop) | Carbon monoxide | 80% | Sharma et al. [91] | |
| N-doped carbon wrapped CNT | 0.5–0.1 | −0.5 V vs. RHE | Carbon monoxide | Cui et al. [93] | ||
| N-CNTs/SS | 2 | −1.1 V vs. Ag/AgCl | Carbon monoxide Hydrogen | 75% | Liu et al. [94] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messias, S.; Nunes da Ponte, M.; S. Reis-Machado, A. Carbon Materials as Cathode Constituents for Electrochemical CO2 Reduction—A Review. C 2019, 5, 83. https://doi.org/10.3390/c5040083
Messias S, Nunes da Ponte M, S. Reis-Machado A. Carbon Materials as Cathode Constituents for Electrochemical CO2 Reduction—A Review. C. 2019; 5(4):83. https://doi.org/10.3390/c5040083
Chicago/Turabian StyleMessias, Sofia, Manuel Nunes da Ponte, and Ana S. Reis-Machado. 2019. "Carbon Materials as Cathode Constituents for Electrochemical CO2 Reduction—A Review" C 5, no. 4: 83. https://doi.org/10.3390/c5040083
APA StyleMessias, S., Nunes da Ponte, M., & S. Reis-Machado, A. (2019). Carbon Materials as Cathode Constituents for Electrochemical CO2 Reduction—A Review. C, 5(4), 83. https://doi.org/10.3390/c5040083