Interactions between a H2 Molecule and Carbon Nanostructures: A DFT Study



Abstract
:1. Introduction
2. Computational Methods
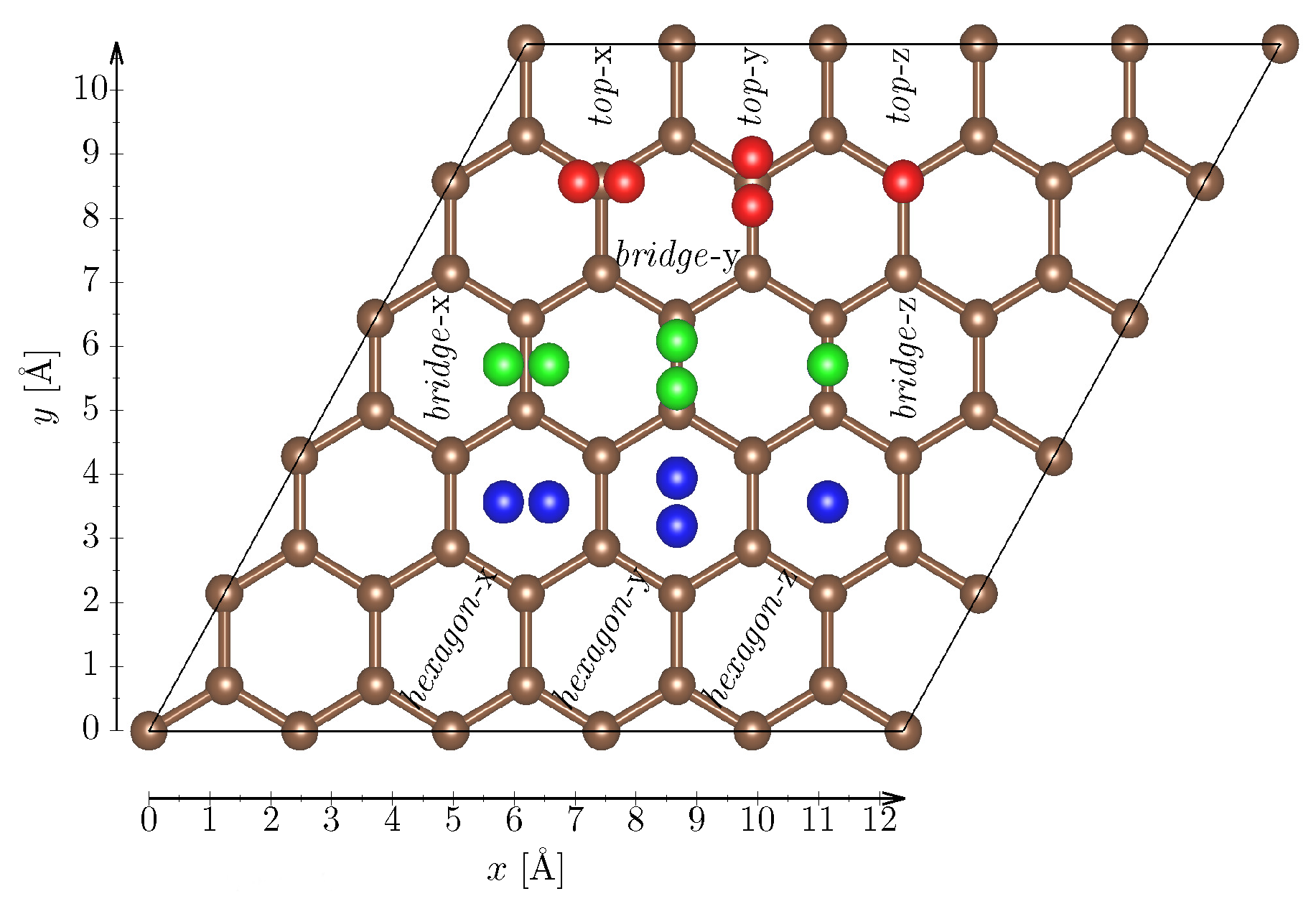
2.1. Graphene Adsorption Curves
- Fully relax the position and geometry of the H molecule, while keeping the C atoms in the graphene plane fixed.
- Vary the height of the molecule with fixed geometry above the graphene sheet ranging from 2 Å to 12 Å.
- Calculate the interaction energy as:
2.2. Adsorption Maps
3. Results
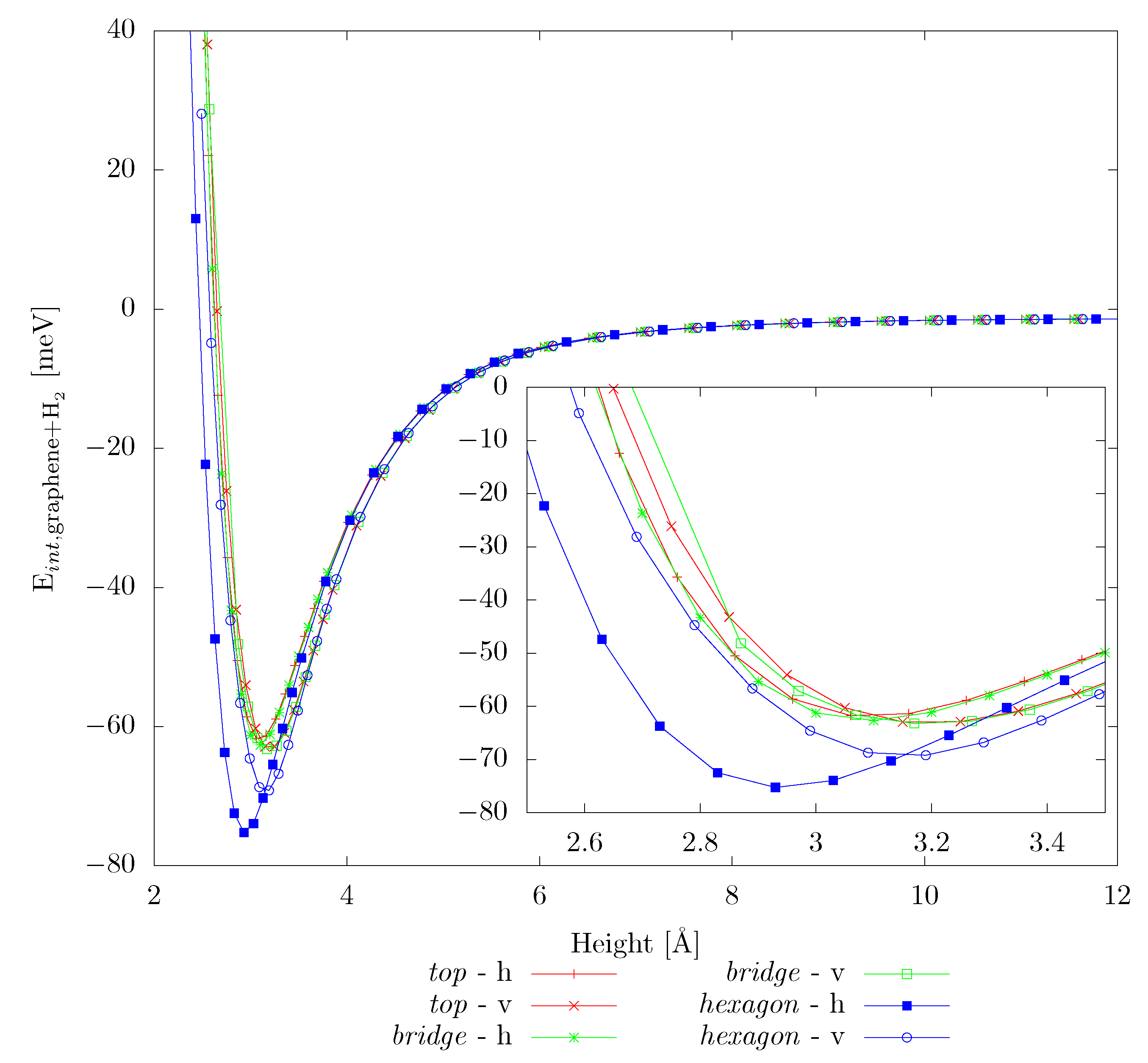
3.1. Hydrogen Adsorption on Pristine Graphene
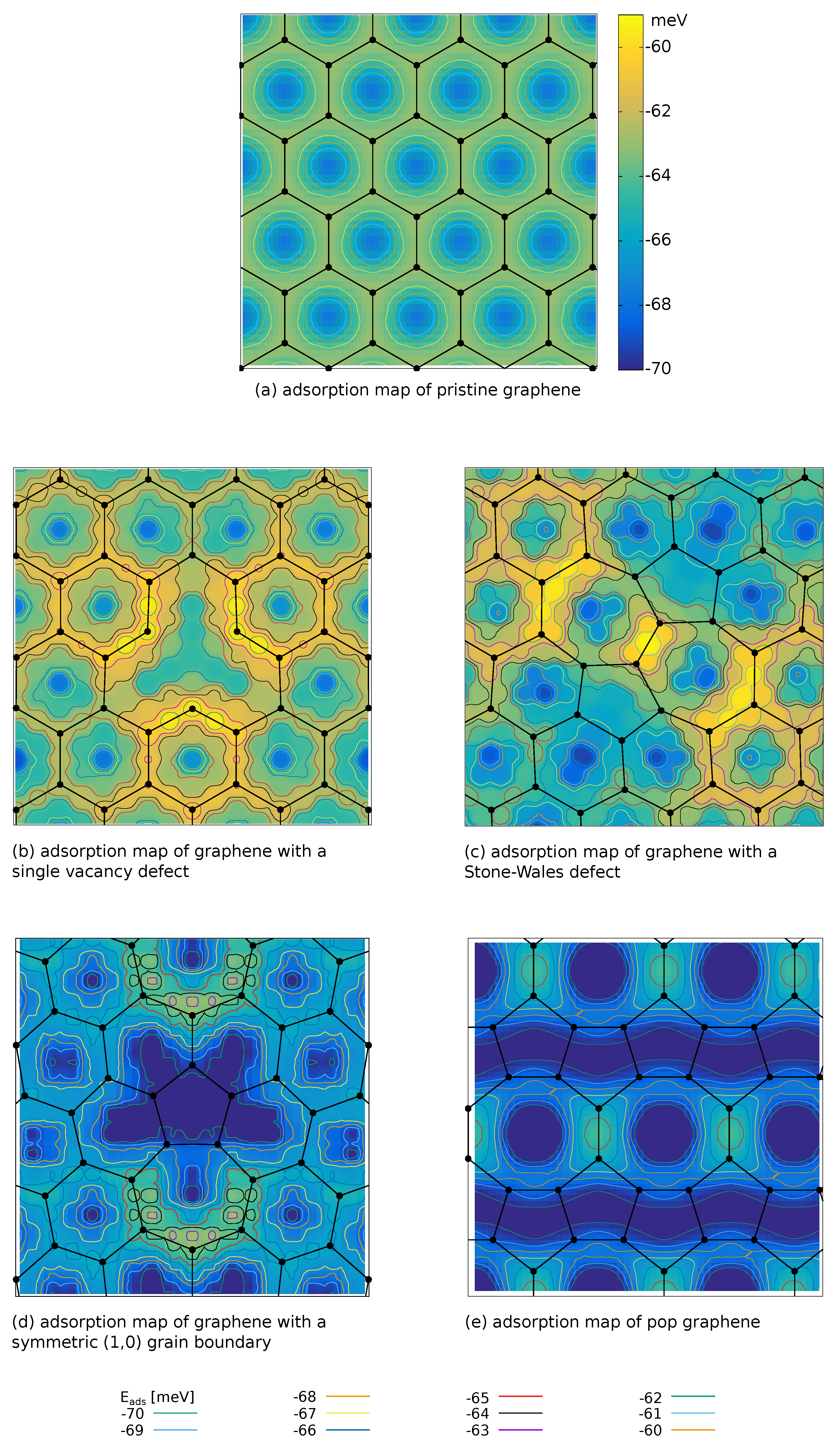
3.2. Adsorption Maps and Pristine Graphene
3.3. Point Defects
3.4. Line Defects
3.5. Globally Modified Graphene
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
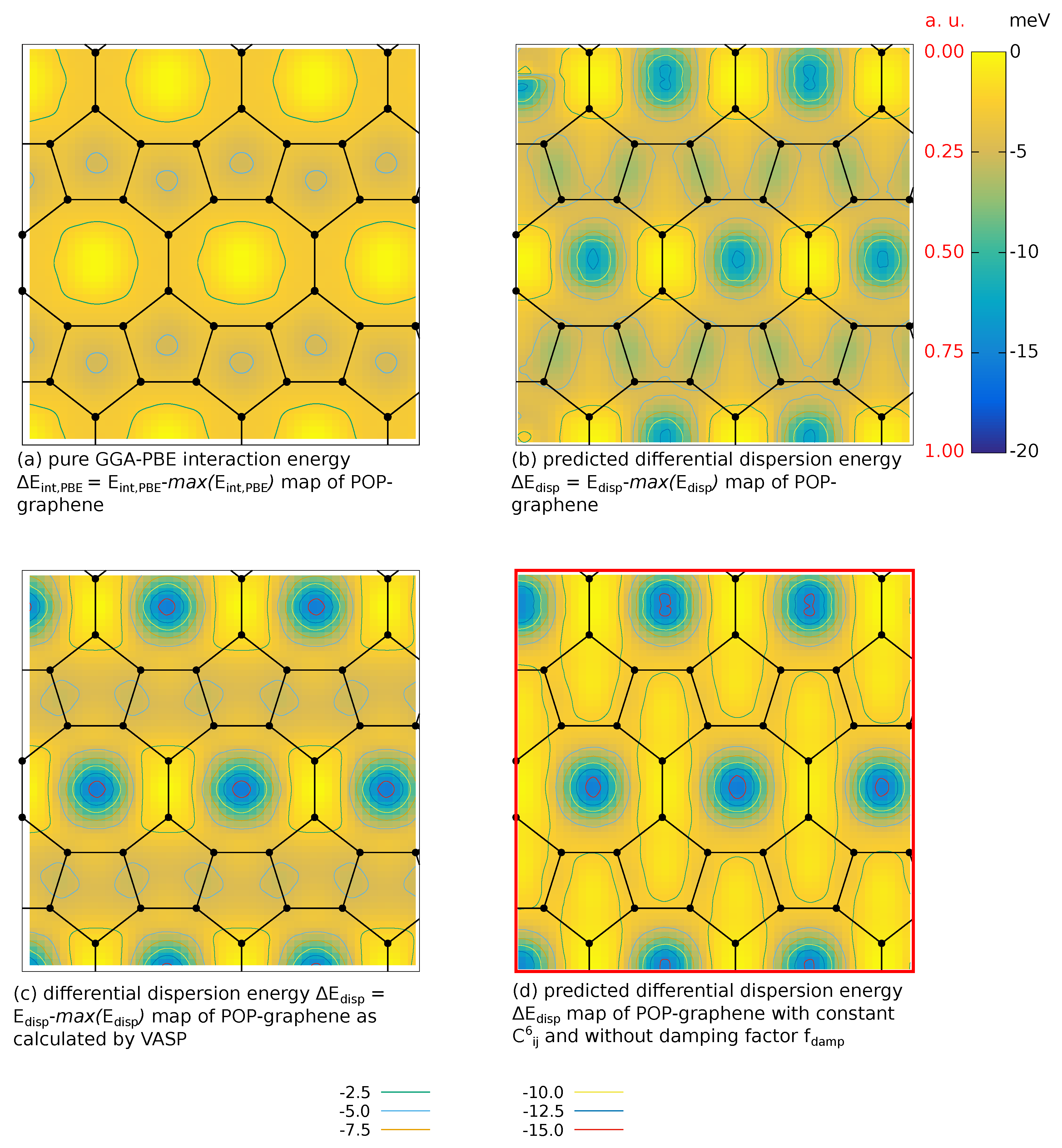
Appendix A. Benchmark Study of Different van-der-Waals Corrections in VASP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Unit | None | D2 | D3 | D3-BJ | TS | TS+IHiPart | MBD | dDsC |
|---|---|---|---|---|---|---|---|---|---|
| IVDW | 0 | 10 | 11 | 12 | 20 | 21 | 202 | 4 | |
| 7.978 | 6.447 | 6.952 | 6.709 | 6.708 | 6.710 | 6.743 | 6.707 | ||
| 1.270 | 0.261 | 0.244 | 0.001 | 0.000 | 0.002 | 0.035 | 0.001 | ||
| % | 18.933 | 3.891 | 3.637 | 0.015 | 0.000 | 0.030 | 0.522 | 0.015 | |
| 2.467 | 2.463 | 2.466 | 2.466 | 2.460 | 2.460 | 2.461 | 2.455 | ||
| 0.006 | 0.002 | 0.005 | 0.005 | 0.001 | 0.001 | 0.000 | 0.006 | ||
| % | 0.244 | 0.081 | 0.203 | 0.203 | 0.041 | 0.041 | 0.000 | 0.244 | |
| 1.270 | 0.261 | 0.244 | 0.005 | 0.001 | 0.002 | 0.035 | 0.006 | ||
| % | 18.934 | 3.892 | 3.643 | 0.204 | 0.041 | 0.050 | 0.522 | 0.244 | |
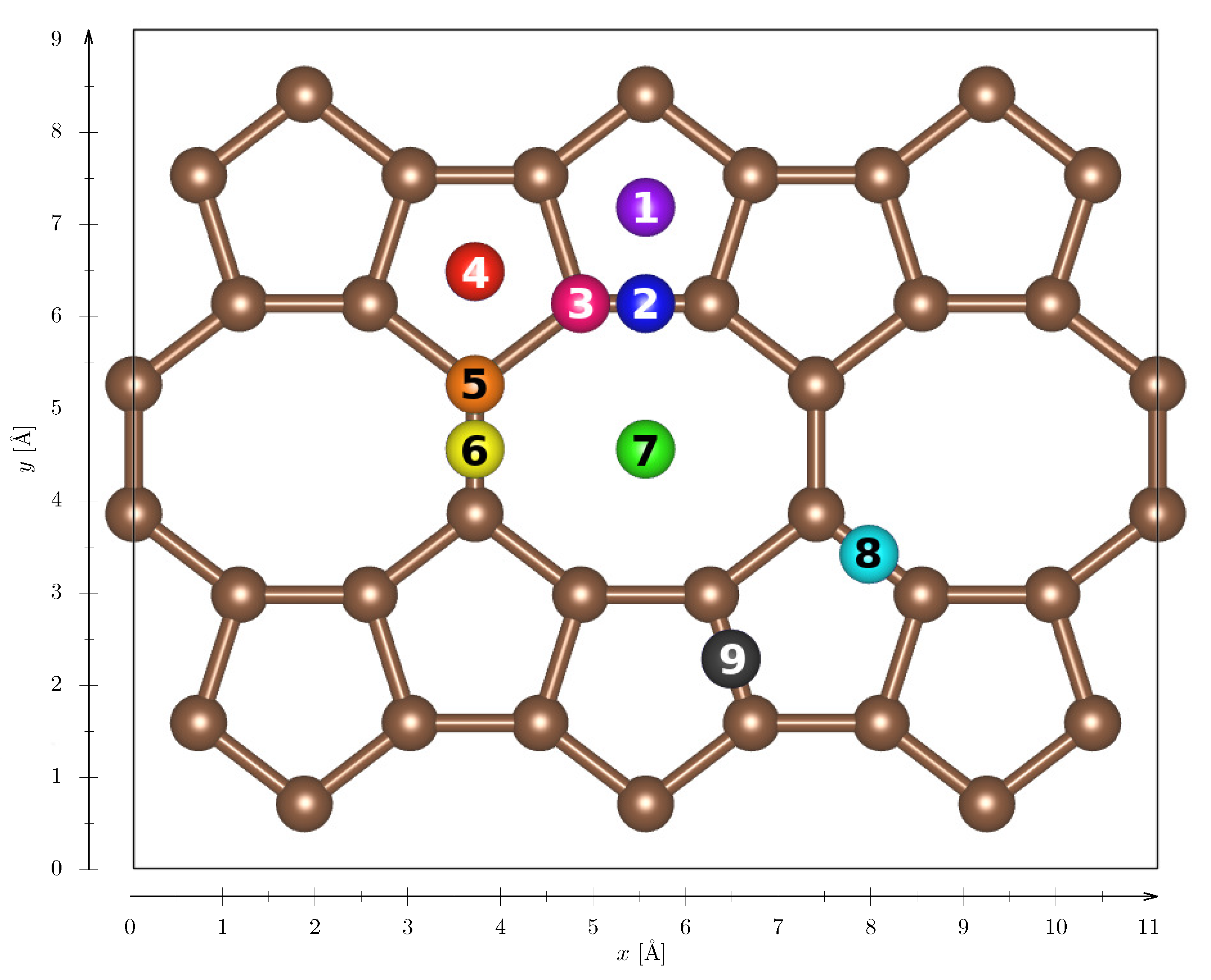
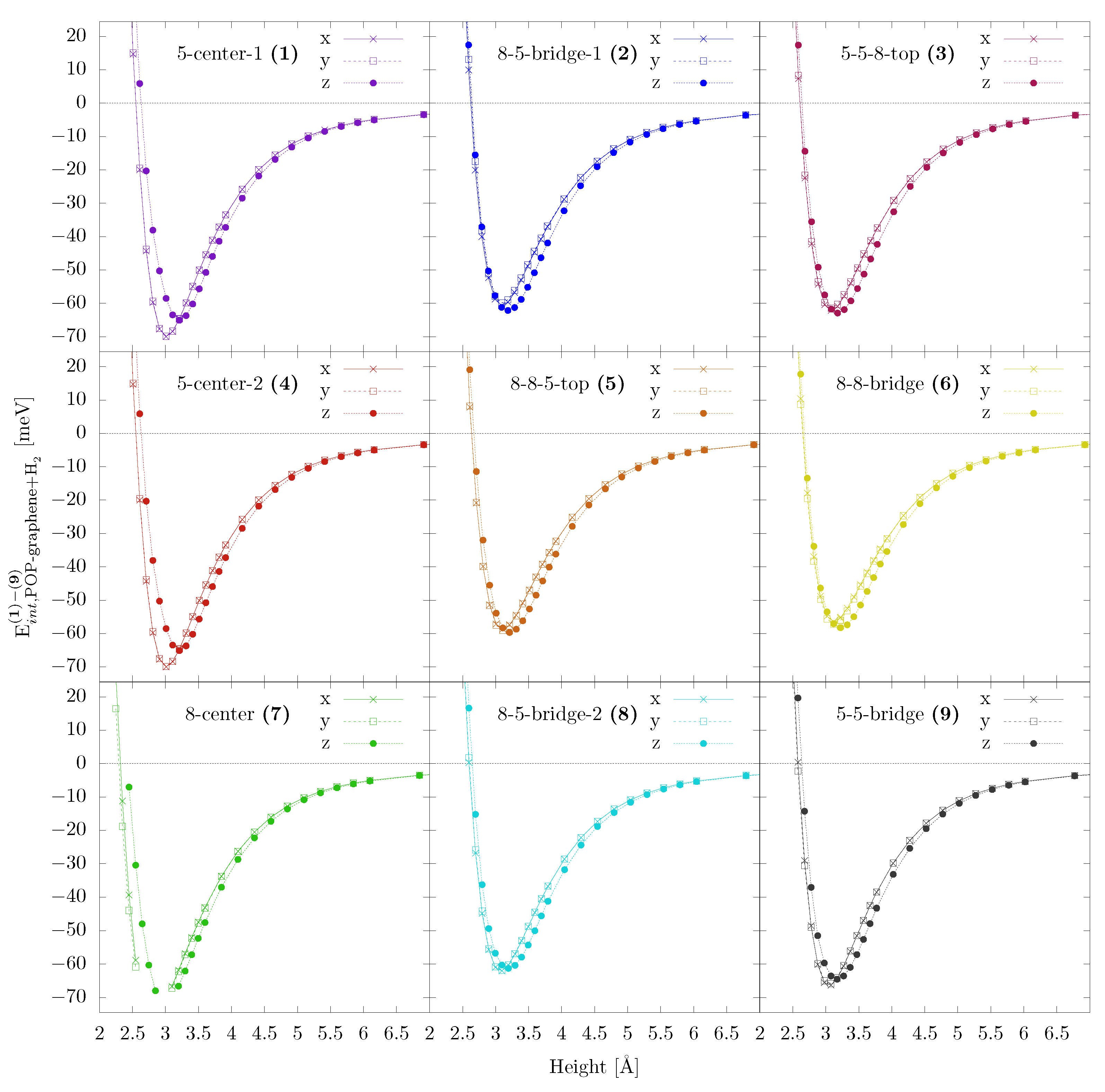
Appendix B. Adsorption POP-graphene


References
- Bastos-Neto, M.; Patzschke, C.; Lange, M.; Möllmer, J.; Möller, A.; Fichtner, S.; Schrage, C.; Lässig, D.; Lincke, J.; Staudt, R.; et al. Assessment of hydrogen storage by physisorption in porous materials. Energy Environ. Sci. 2012, 5, 8294–8303. [Google Scholar] [CrossRef]
- Novoselov, K.S. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Petrushenko, I.K.; Petrushenko, K.B. Hydrogen adsorption on graphene, hexagonal boron nitride, and graphene-like boron nitride-carbon heterostructures: A comparative theoretical study. Int. J. Hydrogen Energy 2018, 43, 801–808. [Google Scholar] [CrossRef]
- Mattera, L.; Rosatelli, F.; Salvo, C.; Tommasini, F.; Valbusa, U.; Vidali, G. Selective adsorption of 1H2 and 2H2 on the (0001) graphite surface. Surf. Sci. 1980, 93, 515–525. [Google Scholar] [CrossRef]
- Rangel, E.; Sansores, E. Theoretical study of hydrogen adsorption on nitrogen doped graphene decorated with palladium clusters. Int. J. Hydrogen Energy 2014, 39, 6558–6566. [Google Scholar] [CrossRef]
- Nguyen, M.T. Reactivity of Graphene and Hexagonal Boron Nitride In-Plane Heterostructures with Oxygen: A DFT Study. ChemPhysChem 2014, 15, 2372–2376. [Google Scholar] [CrossRef]
- Petrushenko, I.K. A DFT Study of Hydrogen Adsorption onto Graphene: Effect of Nitrogen Doping. J. Nano- Electron. Phys. 2017, 9, 03018–1–03018–5. [Google Scholar] [CrossRef] [Green Version]
- Gadipelli, S.; Guo, Z.X. Graphene-based materials: Synthesis and gas sorption, storage and separation. Prog. Mater. Sci. 2015, 69, 1–60. [Google Scholar] [CrossRef] [Green Version]
- Bakhshi, F.; Farhadian, N. Co-doped graphene sheets as a novel adsorbent for hydrogen storage: DFT and DFT-D3 correction dispersion study. Int. J. Hydrogen Energy 2018, 43, 8355–8364. [Google Scholar] [CrossRef]
- Holec, D.; Kostoglou, N.; Tampaxis, C.; Babic, B.; Mitterer, C.; Rebholz, C. Theory-guided metal-decoration of nanoporous carbon for hydrogen storage applications. Surf. Coat. Technol. 2018, 351, 42–49. [Google Scholar] [CrossRef]
- Samantaray, S.S.; Sangeetha, V.; Abinaya, S.; Ramaprabhu, S. Enhanced hydrogen storage performance in Pd3Co decorated nitrogen/boron doped graphene composites. Int. J. Hydrogen Energy 2018, 43, 8018–8025. [Google Scholar] [CrossRef]
- Wu, D.; Yang, B.; Ruckenstein, E.; Chen, H. Functionalization: An Effective Approach to Open and Close Channels for Electron Transfer in Nitrogenated Holey Graphene C2N Anodes in Sodium-Ion Batteries. J. Phys. Chem. Lett. 2019, 10, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Baburin, I.A.; Klechikov, A.; Mercier, G.; Talyzin, A.; Seifert, G. Hydrogen adsorption by perforated graphene. Int. J. Hydrogen Energy 2015, 40, 6594–6599. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yang, B.; Chen, H.; Ruckenstein, E. Reconfiguring graphene for high-performance metal-ion battery anodes. Energy Storage Mater. 2019, 16, 619–624. [Google Scholar] [CrossRef]
- Stone, A.; Wales, D. Theoretical studies of icosahedral C60 and some related species. Chem. Phys. Lett. 1986, 128, 501–503. [Google Scholar] [CrossRef]
- Thrower, P. The study of defects in graphite by transmission electron microscopy. Chem. Phys. Carbon 1969, 5, 217–320. [Google Scholar]
- Yazyev, O.V.; Louie, S.G. Topological defects in graphene: Dislocations and grain boundaries. Phys. Rev. B 2010, 81, 195420. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.H.; Gajewski, G.; Pao, C.W.; Chang, C.C. Structure, energy, and structural transformations of graphene grain boundaries from atomistic simulations. Carbon 2011, 49, 2306–2317. [Google Scholar] [CrossRef]
- Lusk, M.T.; Carr, L. Creation of graphene allotropes using patterned defects. Carbon 2009, 47, 2226–2232. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yakobson, B.I. Cones, Pringles, and Grain Boundary Landscapes in Graphene Topology. Nano Lett. 2010, 10, 2178–2183. [Google Scholar] [CrossRef] [Green Version]
- Ariza, M.; Ortiz, M. Discrete dislocations in graphene. J. Mech. Phys. Solids 2010, 58, 710–734. [Google Scholar] [CrossRef]
- Wang, S.; Yang, B.; Chen, H.; Ruckenstein, E. Popgraphene: A new 2D planar carbon allotrope composed of 5–8–5 carbon rings for high-performance lithium-ion battery anodes from bottom-up programming. J. Mater. Chem. A 2018, 6, 6815–6821. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 6–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.P.; Richards, W.D.; Jain, A.; Hautier, G.; Kocher, M.; Cholia, S.; Gunter, D.; Chevrier, V.L.; Persson, K.A.; Ceder, G. Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis. Comput. Mater. Sci. 2013, 68, 314–319. [Google Scholar] [CrossRef] [Green Version]
- Oliphant, T.E. Python for Scientific Computing. Comput. Sci. Eng. 2007, 9, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Generalized Gradient Approximation Made Simple John. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkatchenko, A.; Distasio, R.A.; Car, R.; Scheffler, M. Accurate and efficient method for many-body van der Waals interactions. Phys. Rev. Lett. 2012, 108, 1–5. [Google Scholar] [CrossRef]
- Bučko, T.; Lebègue, S.; Hafner, J.; Ángyán, J.G. Improved density dependent correction for the description of London dispersion forces. J. Chem. Theory Comput. 2013, 9, 4293–4299. [Google Scholar] [CrossRef]
- Bučko, T.; Lebègue, S.; Hafner, J.; Ángyán, J.G. Tkatchenko-Scheffler van der Waals correction method with and without self-consistent screening applied to solids. Phys. Rev. B 2013, 87. [Google Scholar] [CrossRef]
- Al-Saidi, W.A.; Voora, V.K.; Jordan, K.D. An Assessment of the vdW-TS Method for Extended Systems. J. Chem. Theory Comput. 2012, 8, 1503–1513. [Google Scholar] [CrossRef]
- Rubes, M.; Bludsky, O. DFT/CCSD(T) Investigation of the Interaction of Molecular Hydrogen with Carbon Nanostructures. ChemPhysChem 2009, 10, 1868–1873. [Google Scholar] [CrossRef]
- Okamoto, Y.; Miyamoto, Y. Ab Initio Investigation of Physisorption of Molecular Hydrogen on Planar and Curved Graphenes. J. Phys. Chem. B 2001, 105, 3470–3474. [Google Scholar] [CrossRef]
- Krishnan, S.; Vadapoo, R.; Riley, K.E.; Velev, J.P. Dispersion-corrected density functional theory comparison of hydrogen adsorption on boron-nitride and carbon nanotubes. Phys. Rev. B 2011, 84. [Google Scholar] [CrossRef] [Green Version]
- Matanović, I.; Belof, J.L.; Space, B.; Sillar, K.; Sauer, J.; Eckert, J.; Bačić, Z. Hydrogen adsorbed in a metal organic framework-5: Coupled translation-rotation eigenstates from quantum five-dimensional calculations. J. Chem. Phys. 2012, 137, 014701. [Google Scholar] [CrossRef] [PubMed]
- Ganji, M.D.; Hosseini-khah, S.M.; Amini-tabar, Z. Theoretical insight into hydrogen adsorption onto graphene: A first-principles B3LYP-D3 study. Phys. Chem. Chem. Phys. 2015, 17, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Björkman, T.; Kurasch, S.; Lehtinen, O.; Kotakoski, J.; Yazyev, O.V.; Srivastava, A.; Skakalova, V.; Smet, J.H.; Kaiser, U.; Krasheninnikov, A.V. Defects in bilayer silica and graphene: Common trends in diverse hexagonal two-dimensional systems. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Kotakoski, J.; Meyer, J.C.; Kurasch, S.; Santos-Cottin, D.; Kaiser, U.; Krasheninnikov, A.V. Stone–Wales-type transformations in carbon nanostructures driven by electron irradiation. Phys. Rev. B 2011, 83. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J.; Marsman, M. The VASP Manual - Vaspwiki. Available online: https://www.vasp.at/wiki/index.php/The_VASP_Manual (accessed on 23 March 2020).
- Brinck, T.; Murray, J.S.; Politzer, P. Polarizability and volume. J. Chem. Phys. 1993, 98, 4305–4306. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Chu, X.; Dalgarno, A. Linear response time-dependent density functional theory for van der Waals coefficients. J. Chem. Phys. 2004, 121, 4083–4088. [Google Scholar] [CrossRef]




| System | k-Mesh Type | k-Mesh | Supercell |
|---|---|---|---|
| graphene | -centered | ||
| single vacancy | -centered | ||
| Stone–Wales defect | -centered | ||
| grain boundary | Monkhorst-Pack | ||
| POP-graphene | Monkhorst-Pack |
| System | Supercell Lattice | Sampling Lattice | Irreducible Wedge | Samples | Symmetry Operations |
|---|---|---|---|---|---|
| graphene | Å hexagonal | hexagonal | , | 121 | |
| single vacancy | Å hexagonal | triangle | , | 66 | |
| Stone–Wales defect | Å hexagonal | hexagonal (rotated by 30 with respect to the supercell lattice) | , | 110 | |
| grain boundary | Å, Å orthorhombic | rectangular | , | 170 | |
| POP-graphene | Å, Å orthorhombic | rectangular | , | 285 |
| Configuration | DFT-PBE-TS | DFT-PBE-D3 [4] | |||
|---|---|---|---|---|---|
| Site | Alignment | ||||
| hexagon | v | −69 | 3.15 | −69 | 3.10 |
| bridge | v | −63 | 3.19 | −63 | 3.18 |
| top | v | −62 | 3.19 | −63 | 3.18 |
| hexagon | h | −75 | 2.94 | −67 | 3.10 |
| bridge | h | −63 | 3.09 | −58 | 3.16 |
| top | h | −62 | 3.10 | −57 | 3.16 |
| System | Surface [%] | Increased Surface [%] | |||
|---|---|---|---|---|---|
| min | max | ||||
| graphene | −68.7 | −62.7 | 59.41 | 4.95 | |
| single vacancy | −68.9 | −59.6 | 32.02 | 1.47 | 0.14 |
| Stone–Wales defect | −70.2 | −59.1 | 46.88 | 4.93 | 1.43 |
| grain boundary | −72.6 | −62.4 | 94.09 | 38.48 | 28.23 |
| POP-graphene | −77.4 | −64.2 | 100.00 | 71.39 | 49.91 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gehringer, D.; Dengg, T.; Popov, M.N.; Holec, D. Interactions between a H2 Molecule and Carbon Nanostructures: A DFT Study. C 2020, 6, 16. https://doi.org/10.3390/c6010016
Gehringer D, Dengg T, Popov MN, Holec D. Interactions between a H2 Molecule and Carbon Nanostructures: A DFT Study. C. 2020; 6(1):16. https://doi.org/10.3390/c6010016
Chicago/Turabian StyleGehringer, Dominik, Thomas Dengg, Maxim N. Popov, and David Holec. 2020. "Interactions between a H2 Molecule and Carbon Nanostructures: A DFT Study" C 6, no. 1: 16. https://doi.org/10.3390/c6010016
APA StyleGehringer, D., Dengg, T., Popov, M. N., & Holec, D. (2020). Interactions between a H2 Molecule and Carbon Nanostructures: A DFT Study. C, 6(1), 16. https://doi.org/10.3390/c6010016