
Preparation of Pt/CNT Thin-Film Electrodes by Electrochemical Potential Pulse Deposition for Methanol Oxidation


 , ,
, ,  and
and 
Abstract
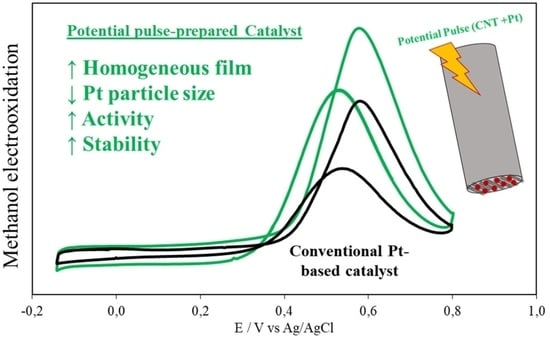
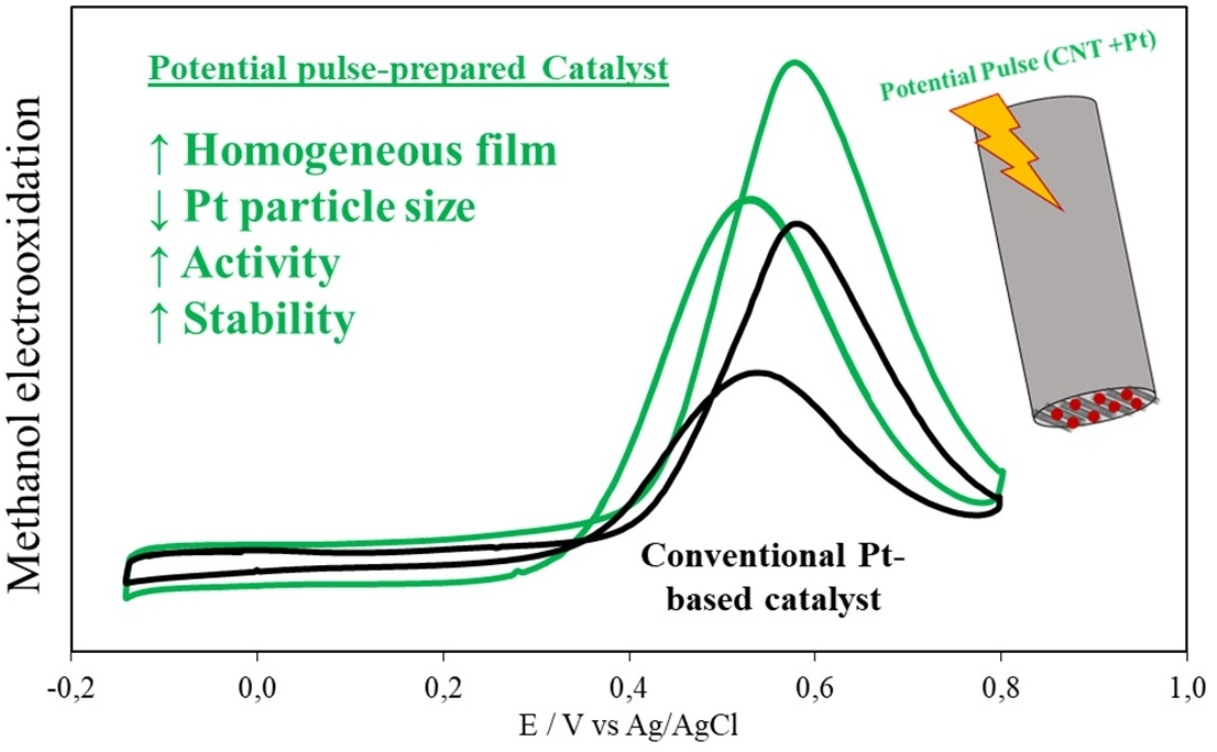
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. CNT Functionalization
2.3. Characterization of the fCNT dispersions
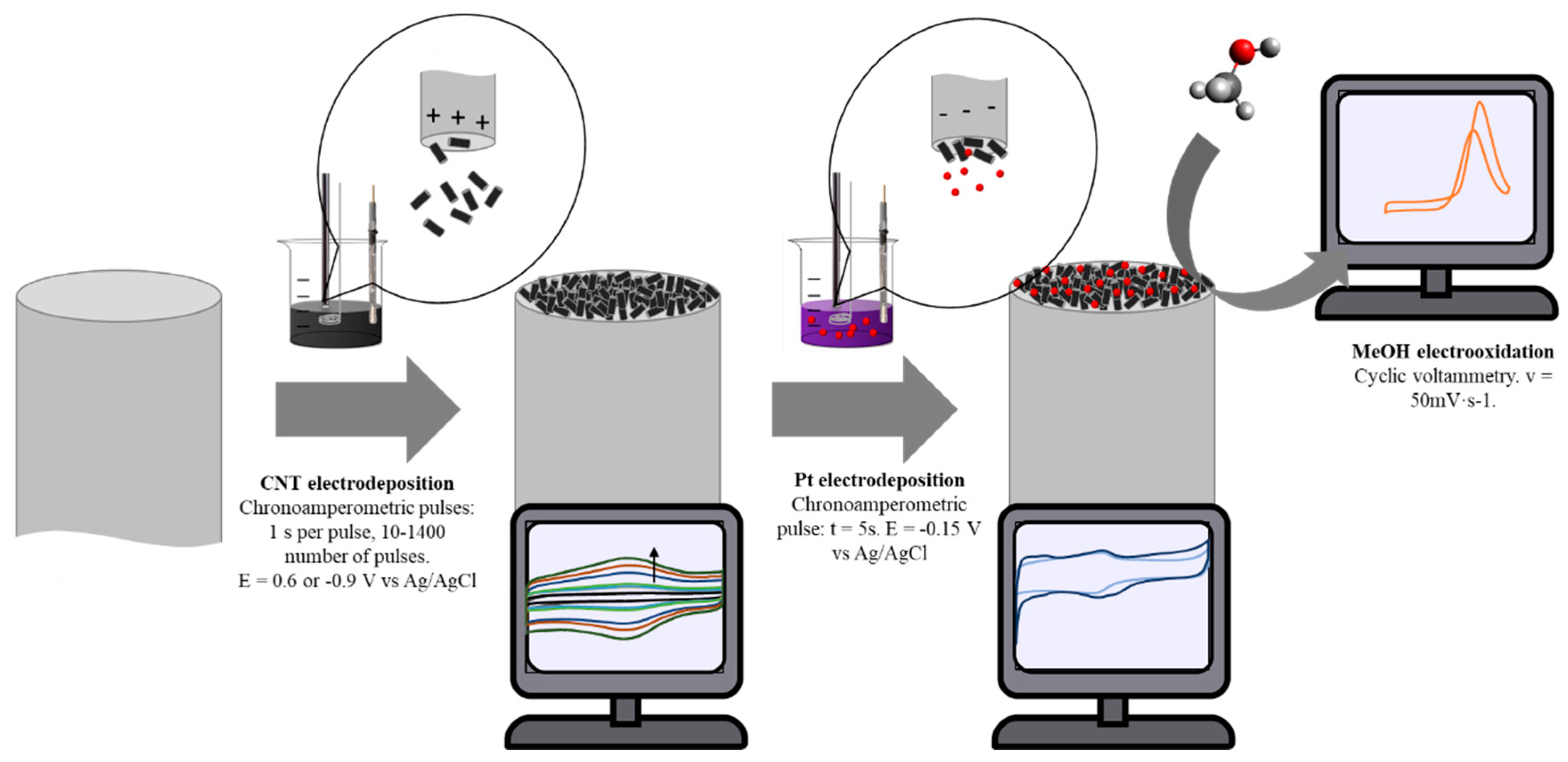
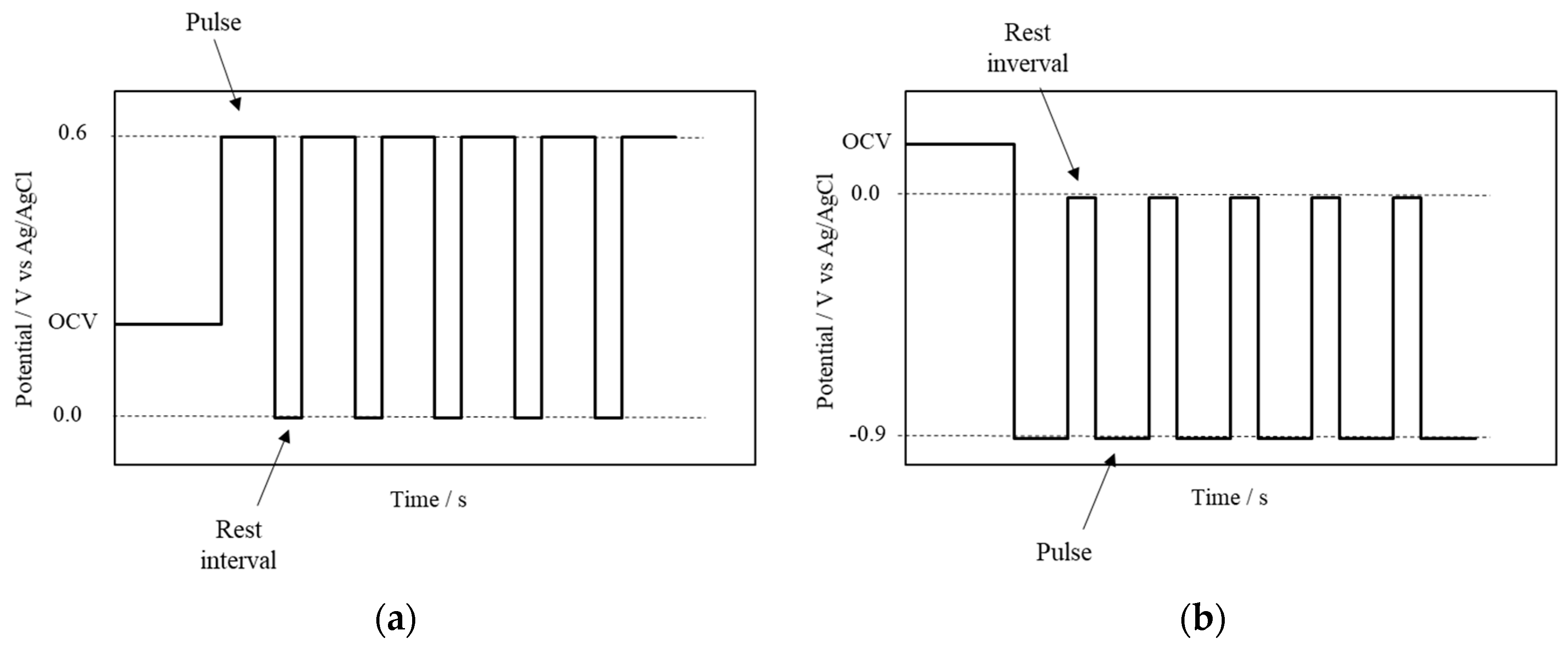
2.4. Electrochemically Assisted Deposition of fCNTs
2.5. Electrochemical Characterization of Electrochemically Assisted Deposited fCNTs
2.6. Electrodeposition of Platinum Nanoparticles
2.7. Physicochemical Characterization
2.8. Electrocatalytic Test towards Methanol Electrooxidation
3. Results and Discussion
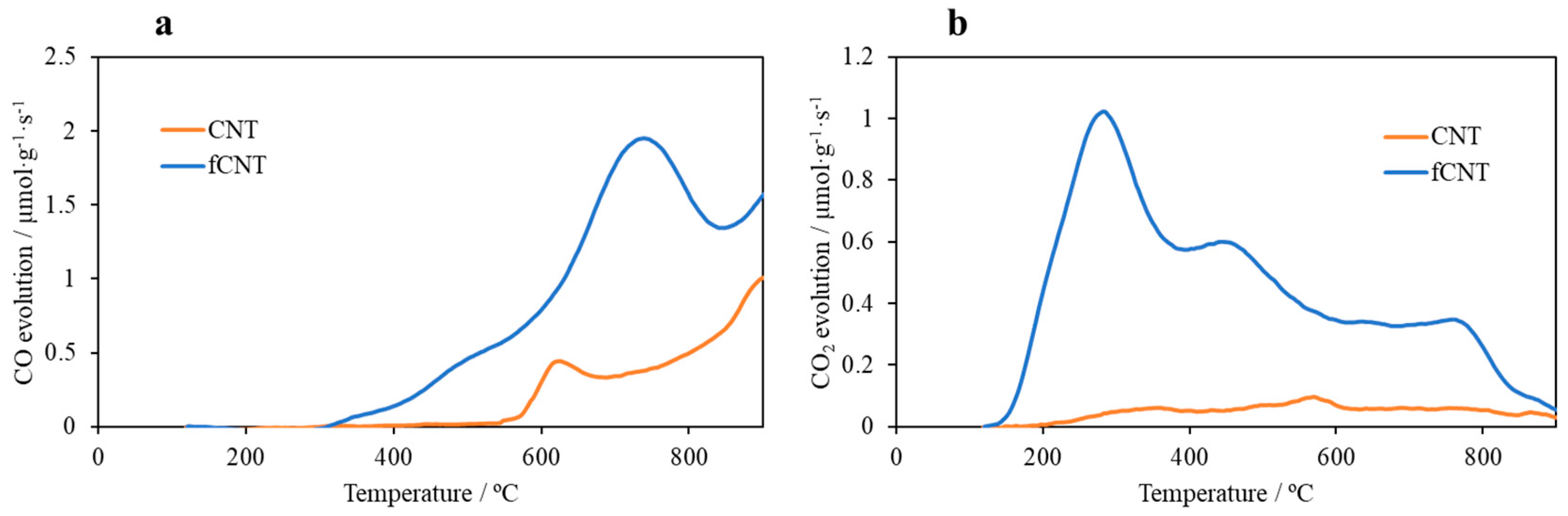
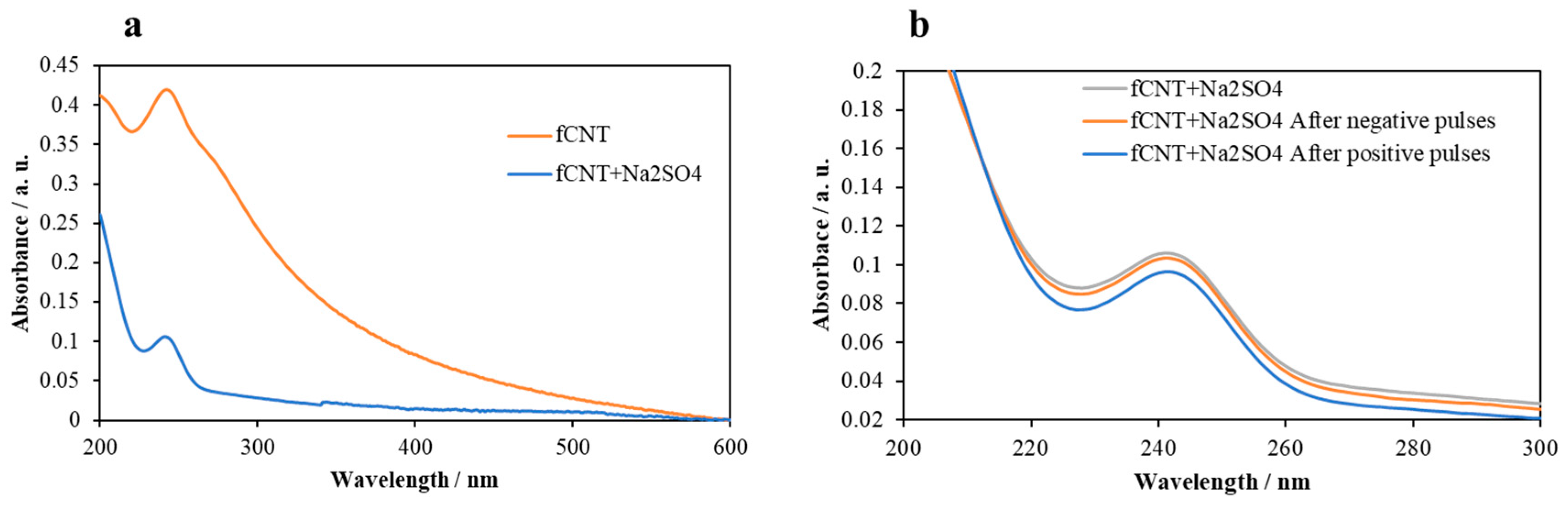
3.1. Characterization of the fCNT dispersions
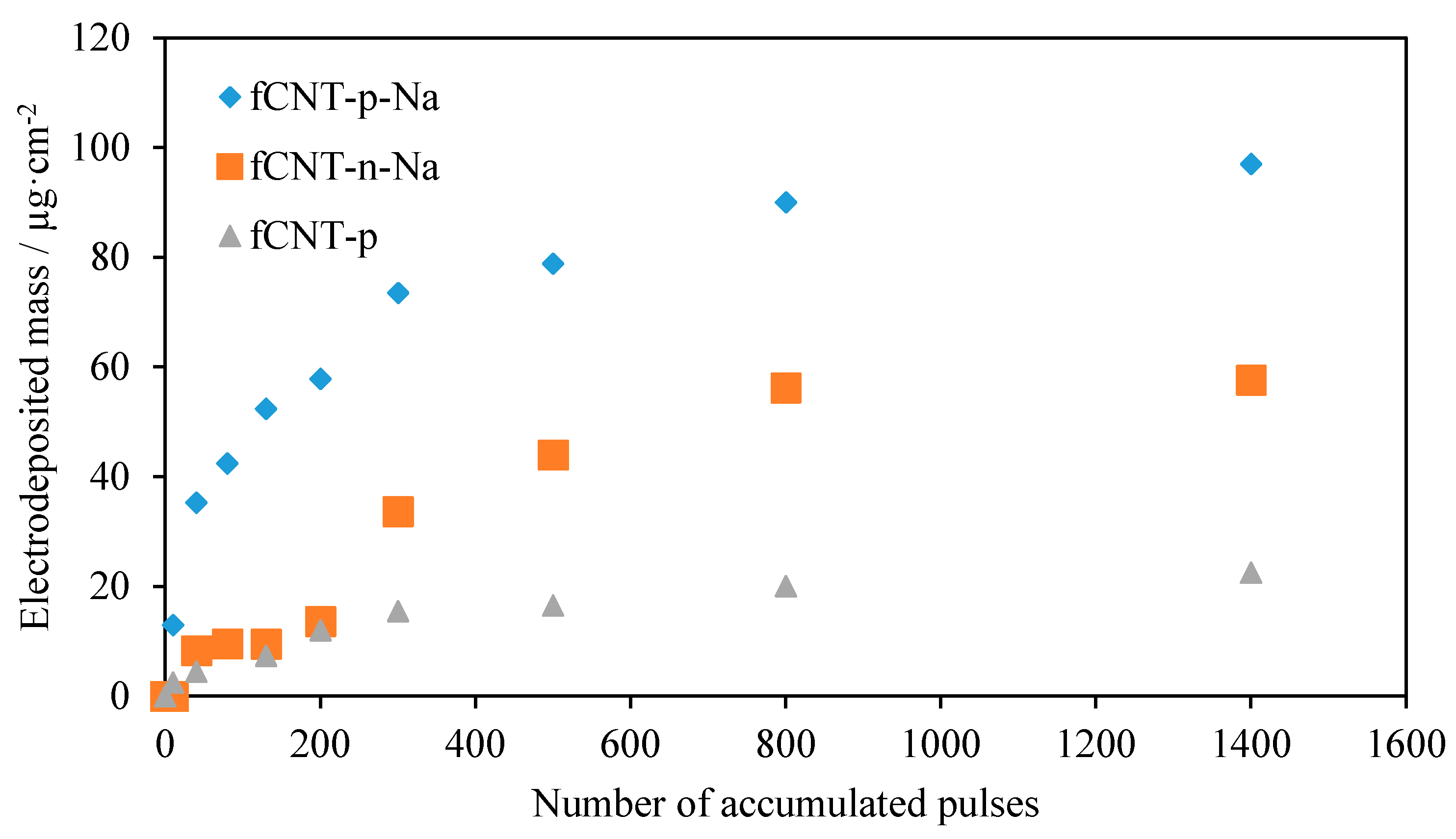
3.2. Electrochemically Assisted Deposition of fCNTs
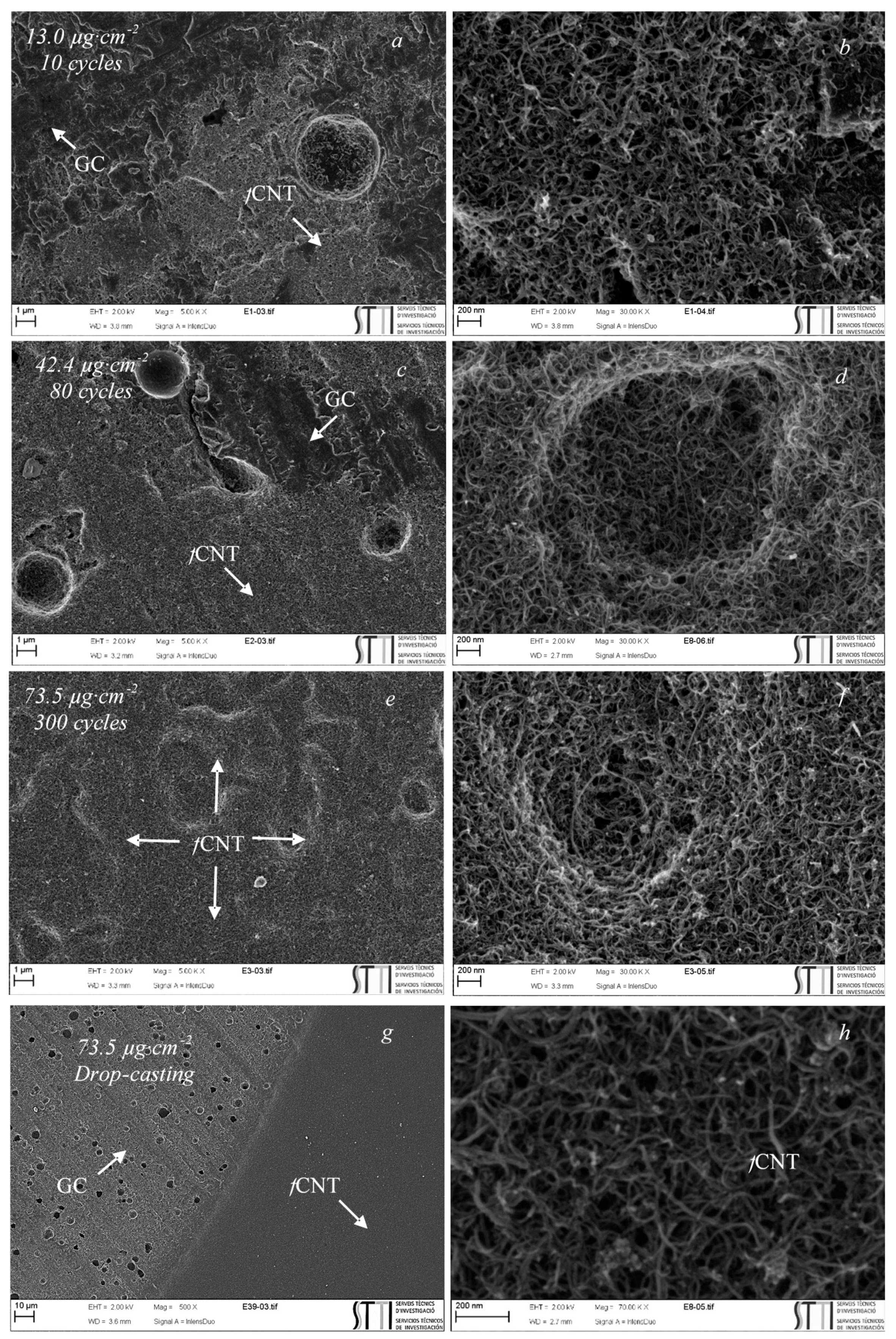
3.3. Physicochemical Characterization of the Electrodeposited fCNTs
3.4. Electrochemical Potential Step Deposition of Platinum Nanoparticles over Electrochemically Assisted Deposited fCNTs
3.5. Electrocatalytic Activity towards MeOH Electrooxidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef]
- Thackeray, M.M.; Wolverton, C.; Isaacs, E.D. Electrical energy storage for transportation- approaching the limits of, and going beyond, lithium-ion batteries. Energy Environ. 2012, 5, 7854–7863. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, J.; Kintner-Meyer, M.C.W.; Lu, X.; Choi, D.; Lemmon, J.P.; Liu, L. Electrochemical Energy Storage for Green Grid. Chem. Rev. 2011, 111, 3577–3613. [Google Scholar] [CrossRef] [PubMed]
- Mccrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef]
- Stephens, I.E.L.; Bondarenko, A.S.; Grønbjerg, U.; Rossmeisl, J.; Chorkendorff, I. Understanding the electrocatalysis of oxygen reduction on platinum and its alloys. Energy Environ. Sci. 2012, 5, 6744–6762. [Google Scholar] [CrossRef] [Green Version]
- Ruqia, B.; Choi, S. Pt and Pt-Ni(OH)2 Electrodes for the Hydrogen Evolution Reaction in Alkaline Electrolytes and Their Nanoscaled Electrocatalysts. ChemSusChem 2018, 11, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.V.; Chen, J.G. Monolayer platinum supported on tungsten carbides as low-cost electrocatalysts: Opportunities and limitations. Energy Environ. Sci. 2011, 4, 3900–3912. [Google Scholar] [CrossRef]
- Ramli, Z.A.C.; Kamarudin, S.K. Platinum-Based Catalysts on Various Carbon Supports and Conducting Polymers for Direct Methanol Fuel Cell Applications: A Review. Nano Rev. 2018, 13, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsounaros, I.; Cherevko, S.; Zeradjanin, A.R.; Mayrhofer, K.J.J. Oxygen Electrochemistry as a Cornerstone for Sustainable Energy Conversion Angewandte. Angew. Chem. Int. Ed. 2014, 53, 102–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Uchaker, E.; Candelaria, S.L.; Cao, G. Nanomaterials for energy conversion and storage. Chem. Soc. Rev. 2013, 42, 3127–3171. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Gu, J.; Su, L.; Cheng, L. An overview of metal oxide materials as electrocatalysts and supports for polymer electrolyte fuel cells. Energy Environ. Sci. 2014, 7, 2535–2558. [Google Scholar] [CrossRef]
- Trogadas, P.; Fuller, T.F.; Strasser, P. Carbon as catalyst and support for electrochemical energy conversion. Carbon 2014, 75, 5–42. [Google Scholar]
- Wang, Y.J.; Zhao, N.; Fang, B.; Li, H.; Bi, X.T.; Wang, H. Carbon-Supported Pt-Based Alloy Electrocatalysts for the Oxygen Reduction Reaction in Polymer Electrolyte Membrane Fuel Cells: Particle Size, Shape, and Composition Manipulation and Their Impact to Activity. Chem. Rev. 2015, 115, 3433–3467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görlin, M.; de Araújo, J.F.; Schmies, H.; Bernsmeier, D.; Dresp, S.; Gliech, M.; Jusys, Z.; Chernev, P.; Kraehnert, R.; Dau, H.; et al. Tracking catalyst redox states and reaction dynamics in Ni-Fe oxyhydroxide oxygen evolution reaction (OER) electrocatalysts: The role of catalyst support and electrolyte pH. J. Am. Chem. Soc. 2017, 139, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Tong, X.; Zhang, G.; Qiao, J.; Gong, Q.; Sun, S. Nitrogen-Doped Carbon Nanotube and Graphene Materials for Oxygen Reduction Reactions. Catalysts 2015, 5, 1574–1602. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Xu, J.; Wei, Z.; Ma, J.; Guo, S.; Wang, S.; Liu, H.; Dou, S. Metal-Free Carbon Materials for CO2 Electrochemical Reduction. Adv. Mater. 2017, 29, 1701784. [Google Scholar] [CrossRef] [PubMed]
- Quílez-Bermejo, J.; Morallón, E.; Cazorla-Amorós, D. Metal-Free Heteroatom-doped Carbon-based catalysts for ORR. A critical assessment about the role of heteroatoms. Carbon 2020, 165, 434–454. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X.; Zhou, Z. Carbon-Based Substrates for Highly Dispersed Nanoparticle and Even Single-Atom Electrocatalysts. Adv. Sci. News 2019, 3, 1900050. [Google Scholar] [CrossRef]
- Kopec, M.; Lamson, M.; Yuan, R.; Tang, C.; Kruk, M.; Zhong, M.; Matyjaszewski, K.; Kowalewski, T. Polyacrylonitrile-derived nanostructured carbon materials. Prog. Polym. Sci. 2019, 92, 89–134. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R. Synthesis and functionalization of carbon xerogels to be used as supports for fuel cell catalysts. J. Energy Chem. 2013, 22, 195–201. [Google Scholar] [CrossRef]
- Ervina, J.; Ghaleb, Z.A.; Hamdan, S.; Mariatti, M. Colloidal Stability of Water-based Carbon Nanotube Suspensions in Electrophoretic Deposition Process: Effect of Applied Voltage and Deposition Time. Compos. Part A Appl. Sci. Manuf. 2019, 117, 1–10. [Google Scholar] [CrossRef]
- Nie, C.; Pan, L.; Li, H.; Chen, T.; Lu, T.; Sun, Z. Electrophoretic deposition of carbon nanotubes film electrodes for capacitive deionization. J. Electroanal. Chem 2012, 666, 85–88. [Google Scholar] [CrossRef]
- Besra, L.; Liu, M. A review on fundamentals and applications of electrophoretic deposition (EPD). Prog. Mater. Sci. 2007, 52, 1–61. [Google Scholar] [CrossRef]
- Haghbin, A.; Liaghat, G.; Hadavina, H.; Arabi, A.M.; Pol, M.H. Enhancement of the Electrical Conductivity and Interlaminar Shear Strength of CNT/GFRP Hierarchical Composite Using an Electrophoretic Deposition Technique. Materials 2017, 10, 1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccaccini, A.R.; Cho, J.; Roether, J.A.; Thomas, B.J.C.; Minay, E.J.; Shaffer, M.S.P. Electrophoretic deposition of carbon nanotubes. Carbon 2006, 44, 3149–3160. [Google Scholar] [CrossRef]
- Haghbin, A.; Liaghat, G.H.; Arabi, A.M.; Hadavinia, H.; Pol, M.H. Investigations on electrophoretic deposition of carbon nanotubes on glass textures to improve polymeric composites interface. Compos. Sci. Technol. 2018, 155, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Battisti, A.; los Ojos, D.E.; Ghisleni, R.; Brunner, A.J. Single fiber push-out characterization of interfacial properties of hierarchical CNT-carbon fiber composites prepared by electrophoretic deposition. Compos. Sci. Technol. 2014, 95, 121–127. [Google Scholar] [CrossRef]
- Guo, J.; Lu, C. Continuous preparation of multiscale reinforcement by electrophoretic deposition of carbon nanotubes onto carbon fiber tows. Carbon 2012, 50, 3101–3103. [Google Scholar] [CrossRef]
- Kamat, P.V.; Thomas, K.G.; Barazzouk, S.; Girishkumar, G.; Vinodgopal, K.; Meisel, D. Self-Assembled Linear Bundles of Single Wall Carbon Nanotubes and Their Alignment and Deposition as a Film in a dc Field. J. Am. Chem. Soc. 2004, 126, 10757–10762. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, K.S.; Mishler, J.; Cho, S.C.; Adroher, X.C. A review of polymer electrolyte membrane fuel cells: Technology, applications, and needs on fundamental research. Appl. Energy 2011, 88, 981–1007. [Google Scholar] [CrossRef] [Green Version]
- Berenguer-Murcia, A.; Morallón, E.; Cazorla-Amorós, D.; Linares-Solano, Á. Preparation of thin silicalite-1 layers on carbon materials by electrochemical methods. Microporous Mesoporous Mater. 2003, 66, 331–340. [Google Scholar] [CrossRef]
- Sanchís, C.; Ruiz-Rosas, R.; Berenguer-Murcia, Á.; Morallón, E.; Cazorla-Amorós, D. Switchable surfactant-assisted carbon nanotube coatings: Innovation through pH shift. Front. Mater. 2015, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sanchís, C.; Berenguer-Murcia, Á.; Ruiz-Rosas, R.; Morallón, E.; Cazorla-Amorós, D. Preparation of homogeneous CNT coatings in insulating capillary tubes by an innovative electrochemically-assisted method. Carbon 2014, 67, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Arias-Pardilla, J.; Berenguer-Murcia, Á.; Cazorla-Amóros, D.; Morallón, E. Electrochemical Preparation of Nanoparticle Deposits: Application to Membranes and Catalysis. Membr. Membr. React. Prep. Optim. Sel. 2011, 395–407. [Google Scholar] [CrossRef]
- Dominguez-Dominguez, S.; Arias-Pardilla, J.; Berenguer-Murcia, A.; Morallón, E.; Cazorla-Amorós, D. Electrochemical deposition of platinum nanoparticles on different carbon supports and conducting polymers. J. Appl. Electrochem. 2008, 38, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Ourari, A.; Zerdoumi, R.; Ruiz-Rosas, R.; Morallon, E. Synthesis and Catalytic Properties of Modified Electrodes by Pulsed Electrodeposition of Pt/PANI Nanocomposite. Materials 2019, 12, 723. [Google Scholar] [CrossRef] [Green Version]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Eswaramoorthy, M.; Sen, R.; Rao, C.N.R. A study of micropores in single-walled carbon nanotubes by the adsorption of gases and vapors. Chem. Phys. Lett. 1999, 304, 207–210. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Otake, Y.; Jenkins, R.G. Characterization of oxygen-containing surface complexes created on a microporous carbon by air and nitric acid treatment. Carbon 1993, 31, 109–121. [Google Scholar] [CrossRef]
- Román-Martínez, M.C.C.; Cazorla-Amorós, D.; Linares-Solano, A.; de Lecea, C.S.M. TPD and TPR characterization of carbonaceous supports and Pt/C catalysts. Carbon 1993, 31, 895–902. [Google Scholar] [CrossRef]
- Li, N.; Ma, X.; Zha, Q.; Kim, K.; Chen, Y.; Song, C. Maximizing the number of oxygen.containing functional groups on activated carbon by using ammonium persulfate and improving the temperature-programmed desorption characterization of carbon surface chemistry. Carbon 2011, 49, 5002–5013. [Google Scholar] [CrossRef]
- Gabe, A.; García-Aguilar, J.; Berenguer-Murcia, Á.; Morallón, E.; Cazorla-Amorós, D. Key factors improving oxygen reduction reaction activity in cobalt nanoparticles modified carbon nanotubes. Appl. Catal. B Environ. 2017, 217, 303–312. [Google Scholar] [CrossRef] [Green Version]
- González-Gaitán, C.; Ruiz-Rosas, R.; Morallón, E.; Cazorla-Amorós, D. Electrochemical Methods to Functionalize Carbon Materials. In Chemical Functionalization of Carbon Nanomaterials; Taylor & Francis: Boca Raton, FL, USA, 2015; pp. 256–287. [Google Scholar]
- Moreno-Castilla, C. Adsorption of organic molecules from aqueous solutions on carbon materials. Carbon 2004, 42, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Newcombe, G.; Drikas, M. Adsorption of NOM onto activated carbon: Electrostatic and non-electrostatic effects. Carbon 1997, 35, 1239–1250. [Google Scholar] [CrossRef]
- Sieben, J.M.; Ansón-Casaos, A.; Martínez, M.T.; Morallón, E. Single-walled carbon nanotube buckypapers as electrocatalyst supports for methanol oxidation. J. Power Sources 2013, 242, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ding, Y.; Chen, L.; Luo, S. ScienceDirect Hierarchical reduced graphene oxide supported dealloyed platinum-copper nanoparticles for highly efficient methanol electrooxidation. Int. J. Hydrogen Energy 2017, 42, 6705–6712. [Google Scholar] [CrossRef]
- Li, K.; Xiao, M.; Jin, Z.; Zhu, J.; Ge, J.; Liu, C.; Xing, W. Advanced architecture carbon with in-situ embedded ultrafine titanium dioxide as outstanding support material for platinum catalysts towards methanol electrooxidation. Electrochim. Acta 2017, 235, 508–518. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Liu, X.; Lu, J.; Zhang, G. Promoting Influence of Activated Carbon used in Carbon Paste Electrode on Platinum Nanoparticles Efficiency in Methanol Electrooxidation. Electrochim. Acta 2017, 242, 165–172. [Google Scholar] [CrossRef]
- Sevilla, M.; Sanchís, C.; Valdés-Solís, T.; Morallón, E.; Fuertes, A.B. Highly dispersed platinum nanoparticles on carbon nanocoils and their electrocatalytic performance for fuel cell reactions. Electrochim. Acta 2009, 54, 2234–2238. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
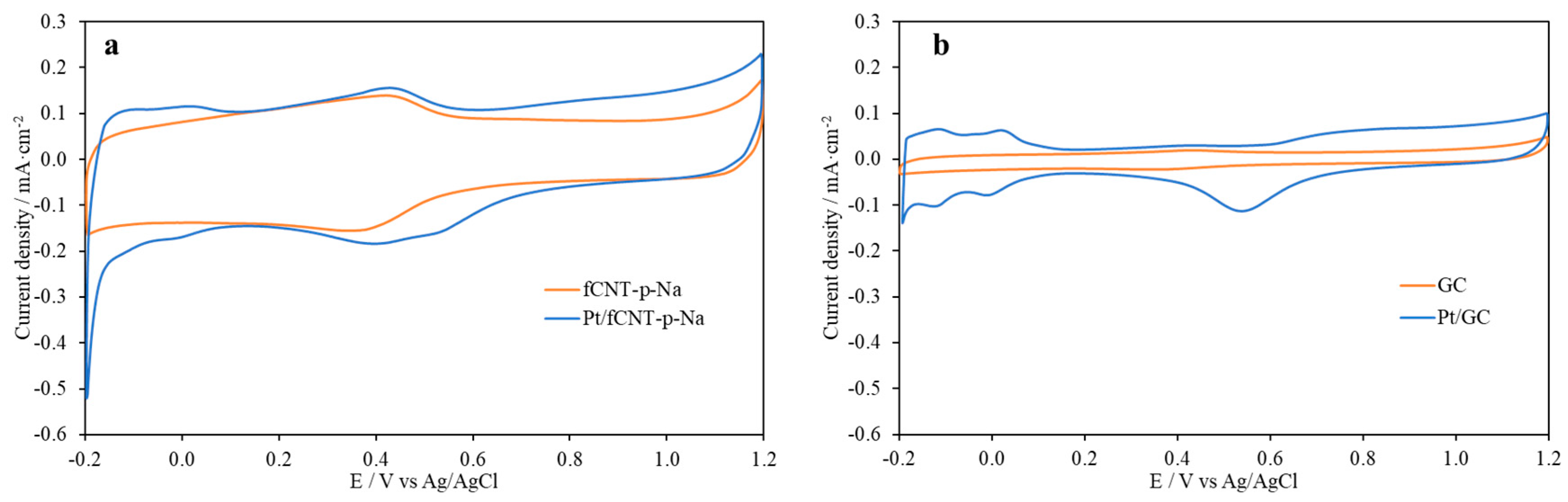{kind=link}
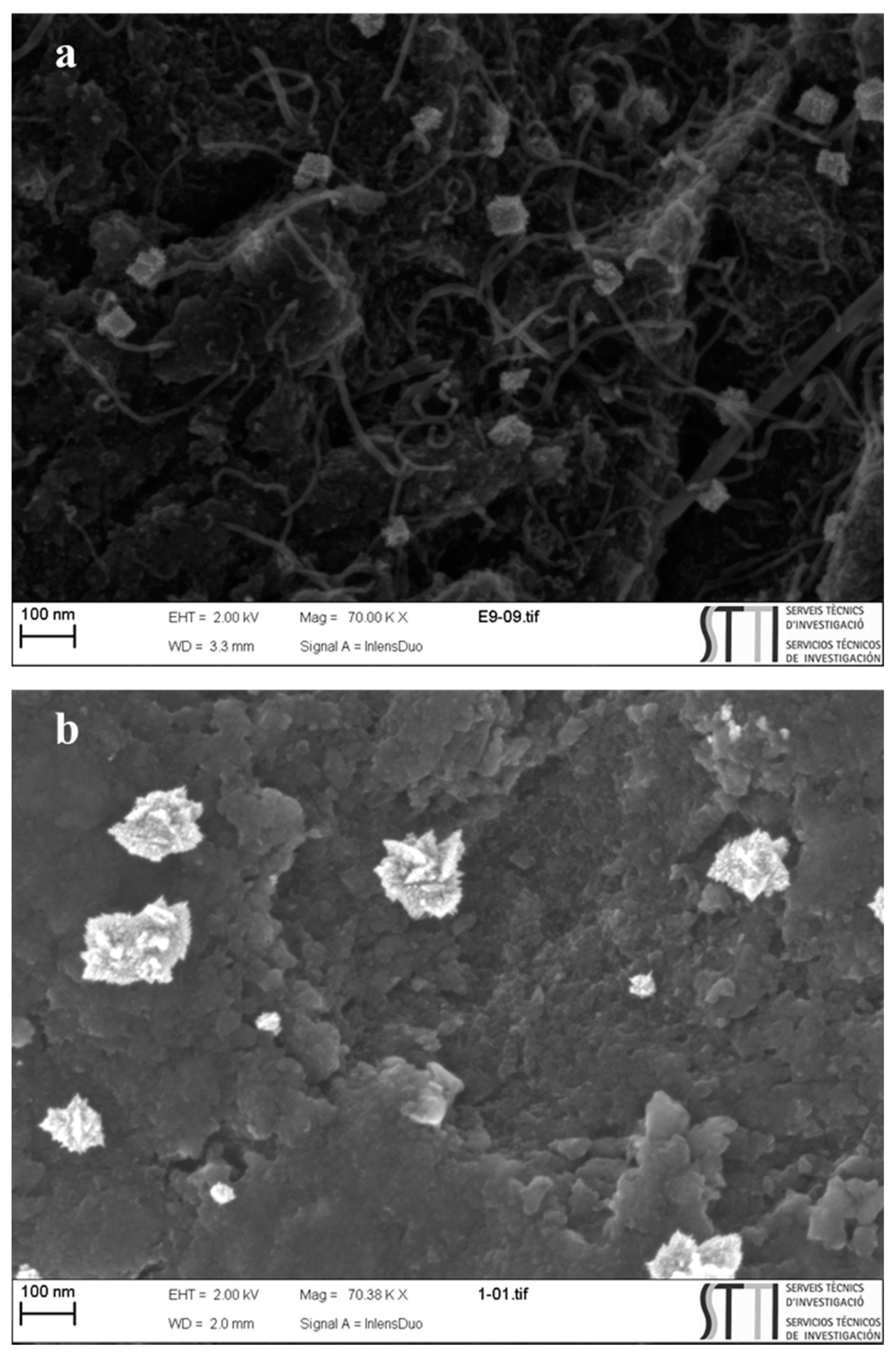{kind=link}
{kind=link}
| Sample | SBET/ m2 g−1 | Vμpore/ cm3 g−1 | CO/ μmol g−1 | CO2/ μmol g−1 |
|---|---|---|---|---|
| CNTs | 260 | 0.01 | 600 | 120 |
| ƒCNTs | 280 | 0.03 | 1740 | 1030 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quintero-Ruiz, J.; Ruiz-Rosas, R.; Quílez-Bermejo, J.; Salinas-Torres, D.; Cazorla-Amorós, D.; Morallón, E. Preparation of Pt/CNT Thin-Film Electrodes by Electrochemical Potential Pulse Deposition for Methanol Oxidation. C 2021, 7, 32. https://doi.org/10.3390/c7020032
Quintero-Ruiz J, Ruiz-Rosas R, Quílez-Bermejo J, Salinas-Torres D, Cazorla-Amorós D, Morallón E. Preparation of Pt/CNT Thin-Film Electrodes by Electrochemical Potential Pulse Deposition for Methanol Oxidation. C. 2021; 7(2):32. https://doi.org/10.3390/c7020032
Chicago/Turabian StyleQuintero-Ruiz, Jose, Ramiro Ruiz-Rosas, Javier Quílez-Bermejo, David Salinas-Torres, Diego Cazorla-Amorós, and Emilia Morallón. 2021. "Preparation of Pt/CNT Thin-Film Electrodes by Electrochemical Potential Pulse Deposition for Methanol Oxidation" C 7, no. 2: 32. https://doi.org/10.3390/c7020032
APA StyleQuintero-Ruiz, J., Ruiz-Rosas, R., Quílez-Bermejo, J., Salinas-Torres, D., Cazorla-Amorós, D., & Morallón, E. (2021). Preparation of Pt/CNT Thin-Film Electrodes by Electrochemical Potential Pulse Deposition for Methanol Oxidation. C, 7(2), 32. https://doi.org/10.3390/c7020032