sp2 Carbon Stable Radicals


Institute of Physical Research and Technology, Peoples’ Friendship University of Russia (RUDN University), 117198 Moscow, Russia
C 2021, 7(2), 31; https://doi.org/10.3390/c7020031
Submission received: 28 January 2021
/
Revised: 4 March 2021
/
Accepted: 9 March 2021
/
Published: 26 March 2021
(This article belongs to the Collection Feature Papers in the Science and Engineering of Carbons)
Abstract
:sp2 Nanocarbons such as fullerenes, carbon nanotubes, and graphene molecules are not only open-shell species, but spatially extended, due to which their chemistry is quite specific. Cogently revealed dependence of the final products composition on size and shape of the carbons in use as well as on the chemical prehistory is accumulated in a particular property—the stabilization of the species’ radical efficiency, thus providing the matter of stable radicals. If the feature is highly restricted and rarely available in ordinary chemistry, in the case of sp2 nanocarbons it is just an ordinary event providing, say, tons-in-mass stable radicals when either producing such widely used technological products as carbon black or dealing with deposits of natural sp2 carbons such as anthracite, shungite carbon, and other. Suggested in the paper is the consideration of stable radicals of sp2 nanocarbons from the standpoint of spin-delocalized topochemistry. Characterized in terms of the total and atomically partitioned number of effectively unpaired electrons as well as of the distribution of the latter over carbon atoms and described by selectively determined barriers of different reactions exhibiting topological essence of intermolecular interaction, sp2 nanocarbons reveal a peculiar topokinetics that lays the foundation of the stability of their radical properties.
1. Introduction
Generally, radicals are objects of the spin chemistry of open-shell molecules. The total pool of molecular spins consists of Nα and Nβ elements, oriented up and down, respectively, and the difference Nα − Nβ formally determines spin multiplicity (SpM) of the molecule ground state. Predominant majority of α and β spins of the molecule inner shells form pairs, both components of which are located in the same space, thus subordinating to the Pauli principle and forming closed-shell spin-orbitals. A part of α and β spins of electrons of outer shells are located in different space, thus forming open-shell spin-orbitals, the same are the relevant molecules. Historically, radical open-shell molecules were attributed to the atomic systems with SpM higher than singlet. However, the open-shell concept is much broader and concerns a much bigger number of the molecule electrons, due to which the spin multiplicity is not the main characteristic of the state and does not exclude the open-shell status of singlet molecule as well. Thus, starting investigation of any new class of molecules, one has to answer two main questions: (i) Are the molecules under study of a closed-shell or open-shell type and (ii) what are the characteristic parameters of the molecule that describe its radical essence. The current paper suggests answers to these questions, related to a family of sp2 nanocarbons. The paper is composed in the following way. A concise historical overview, concerning the spin chemistry of radicals in general, is given in Section 2. Grounds of UHF computational spin chemistry of sp2 nanocarbons are discussed in Section 3. Section 4, Section 5 and Section 6 are devoted, respectively, to spin traits of open-shell molecules of sp2 nanocarbons, electron spins in intermolecular interaction, and the spin nature of the reaction final products, whose post-reaction existence is considered in Section 7. The conclusion summarizes the essences discussed.
2. A Historical Excurse in the Virtual Spin Chemistry of Radicals
The history of quantum-chemical consideration of open-shell molecules is quite long. It originates more than 65 years ago from the paper of Pople and Nesbett [1], who suggested unrestricted Hartree–Fock (UHF) approximation to describe electronic properties of radicals, which has remained conceptually the best until now. First applications of the approach to open-shell species revealed a characteristic UHF feature concerning the spin contamination (SC) of the spin multiplicity of the ground states in the term of the total squared spin profit that is the most pronounced in the singlet case due to . The SC evidences the breaking of spin symmetry of the electronic system. Prominent Lwdin’s papers [2,3] allowed establishing a direct connection between SC and/or broken spin symmetry (BSS) with electron correlation, which results in a concept of configurational interaction (CI). Protesting against the BSS as a physical reality, its appearance in the UHF framework was explained by not taking CI into account to the full extent. From now on, the development of the quantum chemistry of open-shell molecules has continued into two directions. The first stream (CI branch herein) was based on the wish to restore BSS substituting two-determinant UHF computational algorithm by multi-determinant ones that provide CI accounting [3]. Changes concerned wave functions that became more and more complicated while the Hamiltonian of the systems remained unchanged. Many computational schemes has been suggested in this way. A most recent review [4], presented by Chattopadhyay’s team, gives quite complete description of the CI branch modern state of art and supplies readers with many valuable references.
The second UHF branch was based on the acceptance of BSS in open-shell molecules as a physical reality with SC as a characteristic mark of the issue. The approach was originated by Takatsuka, Fueno, and Yamaguchi [5] and was supported by Davidson and his coworkers [6]. Forty-year development of the approach led to its formulation in terms of emergent phenomena in many-electron systems [7,8,9,10,11,12]. Conceptually, this view of electronic systems is as follows. When the many-electron system is spin symmetric, the interaction between electrons adiabatically reduces to the sum of solutions for individual electrons when the interaction falls away. This is typical for closed-shell electron systems and is successfully realized, say, in two Hartree–Fock (HF) versions, making restricted and unrestricted ones identical. When the symmetry is broken, the solution for interacting electrons does not obey this law; as a result, there are new residual (so-called emergent) phenomena at a zero interaction, revealed by UHF [13] and not seen by RHF. Thus, the key question is whether these residual phenomena can be regarded as compatible with physical reality. The answer divides quantum physicists into reductionists and emergentists, saying no and yes, respectively. This view on the situation with open-shell molecules was introduced to the scientific community by the Nobel Prize winner in Physics 1977, P.W. Anderson, in his famous article “More is different” [7], based on the UHF concept. The idea was supported and continued by one more Nobel Prize laureate in Physics 1998, R.B. Laughlin [8], and opened the door to a realm of unexpected phenomena [9,10,11,12]. As it turned out, in physics, emergent phenomena resulted from symmetry breaking are quite numerous and well known. It is enough to mention emergents, inseparable from the solid-state theory such as various quasiparticles: phonons, excitons, solitons, polarons, anions, and so forth, as well as physical phenomena such as topological conductivity, magnetism, and superconductivity. Analyzing the situation with the UHF application to chemistry [14], it turned out to be possible to propose the extension of the emergent concept on chemistry as well, thus addressing the consideration of open-shell molecules in terms of the UHF emergents. Such an approach not only supplies a computationist with classifying markers that make the treatment well-ordered and conceptually self-consistent, but has one more advantage over the CI branch, which is provided with high efficiency of the UHF algorithm, particularly implemented in semi-empirical versions, allowing computational treatment of large molecules.
On practice, in computational spin chemistry of open-shell molecules, there has been a paradoxical situation. Quantum chemists are mostly committed to the reductionist concept of the CI branch. However, the practical application of advanced and most accurate CI methods is still limited to quite small molecular systems, while the real chemistry requires the consideration of large electronic structures containing above a hundred atoms. Meeting these requirements, a lot of effort was made to develop approximate but time-consuming techniques. At the same time, it happened historically that the UHF approximation was publicly repeatedly declared conceptually opposite to the mainstream CI one. Its inherent emergents were heralded to be fundamental errors [15,16] or even unphysical [17]. Expectedly, the approach has been excluded by the mainstream from the proper techniques, and the empty place was quickly occupied with approximate methods, such as density matrix renormalization group (DMRG) [18], restricted active space spin–flip (RAS-SF) [19], and various versions of DFT. If the first two methods could still be considered as some advance towards exact CI methods, then the DFT threw computationists far back into the domain of one-determinant methods. Notwithstanding, the quantum chemical community not only did not resist this seizure, as was in the case of UHF, but encouraged and approved it in every possible way. It did not matter that the DFT in all its aspects not only does not approach CI methods, but is significantly inferior to the UHF approximation. If for closed-shell molecules, this drawback could be compensated by the special empirically based adaptation of functionals, which makes the technique fully empirical, it turned out that DFT was not cut out for open-shell molecules at all (see a detailed discussion of the UDFT problems related to the case [20,21,22]). Similarly to UHF concept, DFT refuses to spin symmetry when transforming into an unrestricted (UDFT) version. In addition, UDFT algorithm operates with spins in non-direct and much more complicated way and touches on not only wavefunctions, but the Hamiltonian as well, thus making the restoration of spin symmetry absolutely impossible [8]. In all the cases of comparative studies, which used UHF, some CI approaches, and UDFT (see a brief review of the issue in [14] as well as the most recent publication [23]), UDFT demonstrated the worth results with respect to the former two techniques, which, in contrast, showed practically identical results. Despite these obvious and well known unfavorable circumstances, over the past two decades, DFT spin chemistry has turned into massive virtual computational chemistry of open-shell molecules, which has flooded hundreds and thousands of scientific publications. Not only UDFT, but widely used restricted DFT as well, can be met on these pages. To date, the capabilities, availability, and effectiveness of DFT methods have been compared with experimental synthetic and analytical procedures, because of which they have become an integral part of practical chemistry.
It had to happen that this period coincided with the time when specific molecules appeared on the stage of modern chemistry-sp2 nanocarbons, including fullerenes, carbon nanotubes, and graphene. All the species contain even number of electrons. Spin multiplicity of the ground state of all of them must begin with a singlet. A whole army of quantum chemists, equipped with the most modern DFT versions, which are in the public domain, rushed to storm new peaks. On the other hand, quasi CI techniques DMRG and RAS SF, efficient enough to treat a large class of open-shell molecules related to polynuclear aromatic hydrocarbons (PAH) (see review [24]), turned out to be inapplicable to sp2 nanocarbons. At the same time, all these molecules, unique in their properties, are quite complex with respect to handling methods. Accordingly, experimental studies have been noticeably lengthened and became more complicated, which led to the dominant contribution of DFT-based calculations to this field of chemistry, markedly dividing it into real and virtual. Therewith, virtual fullerenics and graphenics confidently heralds the explanation and prediction of everything: new properties, new materials, and, most importantly, new applications. However, real chemistry lags behind and does not reveal these new expectations. This discrepancy was especially noticeable during the fulfillment of one of the programs with the largest budget for material science known to date, which is the international program “Graphene Flagship” [25]. Serious complications, met with fulfilling given promises, first led to the appearance of the terms of “good” and “bad” (high-performance and low-performance) graphene [26], and then six years later, the program founders started discussing the dishonesty of graphene material manufacturers [27]. However, the true reason for the problem is the incorrect concept of graphene as a technological material, which was imposed by the unlimited dictate of virtual DFT graphenics. The true properties of graphene turned out to be undisclosed, while presentations of its chemical and physical properties were erroneous. In practice, graphene really behaves as a radicalized object, the spin essence of which the DFT was unable to notice. Even in the latest publication, concerning a seemingly coordinated attitude to the radical properties of small graphene fragments [28], they are considered with an indispensable eye on the DFT. At the same time, remaining in shadow and developing in parallel, the UHF concept of computational graphenics has acquired new evidence of the legitimacy of the approach and the results obtained [29,30,31,32,33,34,35,36,37]. The semi-empirical UHF method has proven to be very effective so that the calculations of, say, fullerene C60 or C70 do not take more than ten minutes on an advanced personal computer, while systems of 200–400 atoms can be considered for one–three days. This allows performing not only single computations, but computational experiments on numerous objects to prove the words of one of the Nobel Prize laureates in chemistry R. Hoffman: “It goes without saying that theory is really of value when it is used to perform numerical experiments that capture a trend. Not numbers, but a trend” [38]. The current paper is devoted to trends in spin chemistry of sp2 nanocarbons revealed by UHF emergents and supported by numerous experimental evidences. The paper presents the issue from the standpoint of a user of computational quantum-chemistry, based on the experience gained during about thirty years.
3. Grounds of UHF Computational Spin Chemistry of sp2 Nanocarbons
3.1. General Remarks
Naturally, the combination of words “spin chemistry” carries an understanding of the special role of electron spins in the ongoing chemical transformations. To make this process visible from the viewpoint of the UHF-spin chemistry, we shall divide it into four conditional stages that present the main issues of practical chemistry:
- Spin traits of the reactants that enter into reactions;
- Spin kind of the intermolecular interaction that controls such reactions;
- Spin nature of the reaction final products;
- Post-reaction storage of spin chemistry products.
Before starting, we have to realize which features of the above issues will be described in terms of UHF emergents. Among many others, we will mainly concentrate on those that are related to the equilibrium ground state, leaving aside continuous symmetry problems caused by spin symmetry breaking [39] that drastically change the appearance of optical electron [35] and vibrational [40] spectra of open-shell molecules. Concerning the ground state, the following features are of interest:
- Ascertainment of the radical status of an open-shell molecule;
- Evaluation of the molecule chemical activity provided with spin density;
- Establishment of the spin density delocalization over the molecule atoms;
- Determination of the delocalization of the chemical activity over molecule, atoms thus presenting its “chemical portrait”;
- Detection of the spin-chemical topology caused by the multi-target character of the molecules.
The following quantities calculated during the computational experiment are the source of the required information. First, there are the molecule total energies , , and , and two differential energies (= − ) and (=) (subscriptions sg, tr, and ST mark singlet and triplet states and the singlet–triplet energy gap, respectively). The second place is occupied by emergent spin characteristics (= − ), SpDtot, and SpDA as well as ND and NDA (SpDtot and ND describe total spin density and total number of effectively unpaired electrons [5,6], while subscript A matches these values related to atom A). A complete set of {sp2C=C bonds} closes the necessary suit of source data. In what follows, the description of the above issues will be carried out on the example of spin topochemistry of a representative set of sp2 nanocarbon molecules, including fullerene C60, a fragment of a single-walled carbon nanotube (SWCNT), and carbon honeycomb-structure compositions, presenting a set of graphene molecules.
3.2. Common Background for the sp2 Nanocarbons Features
Despite the fact that {sp2C=C bonds} pool ends the above list of initial data, it is the network of these bonds, which is the general structural motive of substances as well as the main reason of their peculiarity. The bonds are quite labile and may change the length in the range of 1,326–2,158 Å (see Section 2 of the monograph [37] and Ref. [41]). In this way, they transform from covalent non-radical to fully radicalized bonds. The radicalization extent depends on the current bond length and starts from zero at the critical bond length and drastically grows when the length exceeds Rcrit. The critical value slightly depends on the bond’s surroundings and changes from 1,395 to 1,408 Å when going from ethylene to hexamethylbenzene. The transformation of the bonds from covalent to radicalized ones leads to the transition of a closed-shell electronic structure to an open-shell one. Certainly, we begin the consideration of the chosen nanocarbon molecules from the description of {sp2C=C bonds} distribution.
It is obvious that in the case of extended bond networks, not only individual bond lengths, but also a composition of the bonds in the pool are significant. The latter particularity is seen in Figure 1, where the bond distribution of three sp2 carbon molecules is presented. Inserts in the figure represent equilibrium structures of fullerene C60, a fragment of (4, 4) SWCNT, and right-angle fragment of a flat honeycomb structure with five benzenoid units along both zigzag and armchair edges, (5, 5) NGr molecule herein. As shown in the figure, in all the cases, the bond lengths cover a large region of ~0,3 Å in width. Those with the length over Rcrit make up more than half of the pool. Evidently, the presence of these bonds lays the foundation of the open-shell character of the molecules. However, the generality of the {sp2C=C bonds} distribution of the sp2 molecules in question ends there. Their distribution plottings are quite different and deserve particular consideration.
Fullerene C60. Widely accepted presentation about the molecule local symmetry Ih is supported by closed-shell (RHF) calculations only. The exact symmetry of the molecule in the UHF approach is Ci [39], and the symmetry difference is clearly evident in Figure 1a. It should be noted that a division of bonds into a bilength pattern involving short and long ones remains in both cases. The difference concerns these bonds’ dispersion, which is 1,385 ± 0,0002 and 1,463 ± 0,003 Å for the bond pairs in the RHF solution and 1,391 ± 0,032 and 1,464 ± 0,013 Å in the UHF case. Obviously, the dispersion changes are not too drastic, due to which the symmetry changing in fullerene C60 presents a perfect example of a continuous symmetry when the reduced symmetry, in fact, remains high to a large extent [43]. In the current case, Ci symmetry of C60 includes 95% of Ih [39]. Nevertheless, the deviation from the Ih is definitely revealed experimentally by the appearance of, say, forbidden transitions in optical [35,39] and vibrational (“silent modes”) [40] spectra of the molecule.
It is important to note that the difference in the RHF and/or UHF description of the molecule {sp2C=C bonds} pool concerns not only the change of equilibrium symmetry, but the reaction of the pool to any impact on the molecule. In the case of closed-shell approximation, each individual addition is local and it only slightly disturbs the distribution of the remaining bonds. In contrast, the UHF approach reveals a considerable rearrangement of the whole pool of bonds at each case of the intrusion, thus demonstrating a collective character of the molecule valence electron structure (see detailed discussion of the feature on many examples in [44]).
(4, 4) SWCNT. The bilength bond structure is characteristic for not only fullerene C60, but a series of PAHs and is commonly considered as a characteristic mark of the molecule aromaticity. Accordingly, until now, sometimes a referring to fullerene C60 as aromatic specie can still be found in literature. However, this format of the bond structure strongly violates in the case of C70 and fully disappears in CNTs and graphene molecules. Figure 1b exhibits the picture of {sp2C=C bonds} distribution of the (4, 4) SWCNT fragment determined in two approximations [45]. In contrast to C60, the closed-shell model reveals 10 groups of the bonds with small dispersion within each group and quite large changing between the groups. The open-shell approximation leads to a remarkable ordering of the tube bond structure, decreasing the group number practically by half. Particularly noticeable is the group in the plotting middle covering a continuous change of the length from 1,436 to 1,431 Å, which is related to the tube sidewall. Actually, when the fragment length increases, this group grows as well. Both distributions in Figure 1b also reveal short-length groups related to the tube cap and long-length ones attributed to the tube open end. When the cap is substituted with open end, the short-length bonds disappear from the plotting. This cap–sidewall–end plotting of the {sp2C=C bonds} remains when tubes diameter increases as well as when (n, n) tubes are substituted with (n, m) ones [45]. The reaction of the {sp2C=C bonds} on chemical addition depends on the locality of the latter. In the sidewall region, the response is quite local, while a considerable disturbance is characteristic for caps and open ends.
(5, 5) NGr molecule. A comparative pattern of {sp2C=C bonds} of the closed- and open-shell compositions of the molecule is similar to that one discussed above for the tube with the only difference being related to the absence of characteristic “sidewall” bonds. Extending the molecule size leads to increasing the groups number while smoothing the difference between the groups, which results in a continuous decreasing of bond lengths from ~1,49 to 1,33 Å in the relevant Z→A plottings. One more peculiarity is characteristic for graphene molecules. It concerns the response of the whole {sp2C=C bonds} pool on the chemical addition, which drastically changes the pool distribution at each action (see a detailed discussion of the feature on various examples in [37]).
Besides the features discussed, Figure 1c draws attention to one more exclusive particularity of the molecule. It concerns the extremely small singlet–triplet gap which in the case of (5, 5) NGr constitutes −6.096 kcal/mol. The triplet state is slightly lower than the singlet, which means the two states are strongly mixed. Applying the UHF approach to both states separately, we obtain quite similar {sp2C=C bonds} plottings, which is clearly seen in the figure.
Completing the description of {sp2C=C bonds} pools, we can draw the following conclusions.
- More than half of sp2C=C bonds of sp2 nanocarbons, including fullerenes, carbon nanotubes, and graphene molecules, are longer than critical interatomic distance , exceeding over which leads to the bond radicalization. The feature lays the foundation of the open-shell character of the species electron systems and radical character of the molecules.
- If bilength bond composition is characteristic for the {sp2C=C bonds} pool of fullerene C60, many-length composition is typical for more extended {sp2C=C bonds} networks starting from fullerene C70 and involving CNTs and graphene molecules.
- Application of UHF approach allows disclosing a collective response of {sp2C=C bonds} pool to each act of any chemical addition to the relevant species in all the cases, thus revealing delocalization of the bond distribution disturbance.
4. Spin Traits of Open-Shell Molecules of sp2 Nanocarbons
4.1. Fullerene C60
Having dealt with the {sp2C=C bonds} pool of the chosen molecules, we now turn to data concerning their energies and spin emergents. The collection of the corresponding source data is presented in Table 1. The data evidence convincingly that the considered molecules are of the open-shell type and their chemistry is spin by nature. The first acquaintance with UHF spin chemistry is best to start with C60 fullerene, which has all characteristic emergents of open-shell molecules, on the one hand, and is a real well-known molecule, on the other, which allows addressing experimental data to confirm computationally predicted emergent features. As seen in Table 1, the UHF approach remarkably lowers the energy of the singlet state, which is much lower than the triplet one and constitutes itself as a ground state. The singlet–triplet gap accounts for ~3% of the total energy and is small enough for the spin contamination of the ground state to be reliably fixed. Due to the singlet character of the ground state, the total number of effectively unpaired electrons ND equals to the doubled spin contamination [5,6]. This number is distributed over the molecule atoms in terms of partial numbers NDA, plotting of which over the molecule atoms is shown in Figure 2a in two manners, namely, as histogram presenting Z→A distribution and dotted curve related to the atom numbers in the output file. The first plotting allows distinguishing six clearly seen groups of atoms with 12 atoms of the same NDA value per each group of the first four groups and with six atoms of groups 5 and 6. Atoms of the first group form two hexagons (see configurations of slight blue atoms in inserts in Figure 1a and Figure 2a), atoms of groups 2, 3, and 4 are joined by pairs (red, rose, and green), and dark blue atoms are singles [46]. Figure 2b presents an A→Z spin density distribution that clearly demonstrates antiferromagnetic delocalization of the density over the molecule atoms providing the SpDtotal exact zeroing.
Since NDA represents atomic chemical susceptibility (ACS) [47], the insert images in Figure 1 and Figure 2 portray the chemical reactivity distributed over the molecule atoms from the most reactive (light blue) to the most inactive (dark blue), thus allowing drawing the fundamentals of the C60 spin chemistry. The first feature of the set states that the molecule is chemically active and its molecular chemical susceptibility (MCS) ND constitutes 9.87 e. According to the second echelon, the molecule is a multi-target chemical object, since all its atoms can participate in the chemical reaction with a fractional ACS contribution NDA. Following the third-step element, the molecule enters any chemical action by atoms of the highest ACS related to the largest NDA value. Since NDA distribution over the molecule is spatially peculiar and changes at each reaction step, a chemical reaction, involving fullerene C60, acquires topological features depending on the chemical counteragent, which is the fourth-step element of the C60 spin chemistry. The fifth element concerns the polyderivative character of the reaction between any reagent and fullerene. The sixth one determines the derivative order related to the reaction completion, which is due to continuous decreasing of the molecular chemical susceptibility ND in the course of the reaction until it is worked out up to nil. Evidently, the reaction may be stopped before ND reaching nil due to sterical obstacles.
4.2. (4, 4) SWCNT Fragment
As it turned out, the fundamentals discussed in the previous section govern generally the spin chemistry of not only C60 and higher fullerenes [44], but also other sp2 nanocarbons such as CNTs and graphene molecules. As seen in Table 1, the UHF approximation eliminates the artifact when the RHF singlet state is located much above the triplet one and displaces the singlet downwards. The energy gap is small and makes up 1.3% of the energy of the singlet state. For the current case, the gap is negative, indicating that formally the triplet should be attributed to the ground state. Notwithstanding, the sign of the gap is not steady and changes when either length or diameter of the tube increases. The only steady point is that the gap is small so that singlet and triplet states are mixed. It should be noted that the spin contaminations of both states, and , as well as the total numbers of effectively unpaired electrons, NDsg and NDtr, are considerable and practically the same. The spin-triplet near-degeneracy, which is typical for polyradicoloids with double C=C bonds [23,24], greatly complicates the description of open-shell electronic states [4,21,22], and a proper computational technique has been still absent. Therefore, we will have to limit ourselves to the available capabilities and hold a discussion of the question posed in terms of the UHF consideration of the singlet state.
The description of the singlet (4, 4) SWCNT molecule as an open-shell system is much the same as that of fullerene C60. Plotting in Figure 3 presents the ACS NDA distribution over the molecule atoms. The distribution convincingly tell us that (i) the tube is a multi-target object; (ii) the main reaction ability is concentrated on open-end atoms and these atoms enter any reaction always first; and (iii) fewer active atoms are located in the cap zone and still fewer active atoms are distributed along sidewall. Different from the fullerene case are the extremely high ACS values for open-end atoms. The feature is connected with the fact that ACS of cap and sidewall atoms is determined by the {sp2C=C bonds} pool, due to which their ACS is comparable with that of fullerene atoms, while ACS of open-end atoms are additionally provided by dangling bonds (DBs). Actually, as seen in the figure, saturation of these DBs by monoatomic hydrogens drastically inhibits the end atoms’ reactivity, equalizing it with the sidewall one. The discussed fundamentals govern all general features of the CNT spin chemistry, which are widely confirmed experimentally [48].
4.3. (5, 5) NGr Molecule
As seen in Table 1, everything that was said earlier with respect to the singlet–triplet degeneracy of the CNT states can be readdressed to this graphene molecule. In the latter case, the degeneracy becomes even more pronounced, since the EST gap decreases even more and accounts for 0.4% of the total energy. Expectedly, the sign of EST can easily alternate and, in fact, this is what happens when size, shape, and chemical modification in the area of edge atoms change. Leaving for the future a correct theoretical consideration of open-shell systems with nearly degenerate states, we restrict ourselves below to the discussion of graphene molecules in the framework of the UHF consideration of one of the degenerate states. As has occurred in the current study, the consideration of either singlet or triplet state leads to practically the same results that might be caused by the close equality of and ND data for both states.
A considerable value of MCS ND evidences high chemical activity of the (5, 5) NGr molecule as a whole. Distributed over the singlet molecule atoms, it is presented as an ACS NDA histogram plotting in Figure 4. The spin density distribution is qualitatively identical to that of fullerene C60 in Figure 2b with SpDtotal equal zero [46]. As for NDA plotting shown in the figure, edge atoms dominate due to large contribution providing with DBs. As in the case of CNT, considered previously, their chemical reactivity can be inhibited by monoatomic termination of edge-atom DBs by hydrogens. In this case, the molecule becomes one of known polyradicaloids belonging to peripentacenes [24], the radical essence of which is presented by the curve with dots in Figure 4. Edge atoms mark the zone of the highest reactivity, while the reactivity of those in the basal plane is mainly determined by the {sp2C=C bonds} pool and is at the level of fullerene and CNT sidewall atoms. The molecule as a whole is evidently multi-target prone to polyderivatization. Each step of polyderivatization is accompanied by the redistribution of remaining sp2C=C bonds and, consequently, of NDA values, thus evidencing a collective character of the electron system of the molecule. A drastic difference in chemical reactivity of edge and basal-plane atoms as well as a sharp anisotropy of the spatial structure in the direction normal to the basal plane provide extreme conditions for topological spin chemistry [49]. Taken as a whole, the described chemical behavior of graphene molecules is observed experimentally. Readers can find a number of examples in monograph [37].
All the peculiarities of molecules discussed above are rooted in the spin symmetry breaking. We share the view that the action is resulted from the occurred quantum transition [14] that makes the UHF approach a good basis for the description of ground state, experimental justifications of which were discussed in [14] and have received the additional confirmation just recently [23]. Characterized in terms of UHF emergent, spin traits of the discussed molecules include the following issues. At atomic level, the latter concerns a large set of local spins that lay the foundation of the over-atom delocalization of both spin density and chemical activity. At the molecular level, high molecular chemical susceptibility, evidencing radical character of the molecules, and topological character of chemical reactions are the main issues. At the energy level, small energy gap of alternate sign draws attention to an exceptional feature concerning singlet–triplet near-degeneracy. The issue causes the necessary inclusion of the development of the quantum theory of open-shell electronic systems with pronounced singlet–triplet degeneracy in the agenda of modern quantum chemistry.
5. Electron Spins in Intermolecular Interaction
When one of the partners of a chemical reaction is of the open-shell type, the intermolecular interaction (IMI) between partners considered in the UHF approximation takes spins into account directly, operating with two kinds of determinants related to α and β spin orbitals. The participation of spins in the formation of this interaction is hidden from the outside view, and the correctness of their accounting can be judged only by results concerning, say, final products of the reaction. This aspect will be discussed in detail in the next section. Herein, we shall consider a particular case when spins are evidently involved in the IMI. The matter is that sp2 nanocarbons molecules are not only an open-shell species, but are characterized by exclusive donor–acceptor records. Quite low ionization potentials and high electron affinities allow them behaving both donors and acceptors simultaneously. The issue is of particular importance when concerning IMI [44,50]. Combining both open-shell and donor–acceptor character, IMI leads sometimes to an exciting result. Photodynamic (PD) effect, directly concerning IMI of fullerene C60 and molecular oxygen [51], can be the best example. The effect is widely used in the medicinal chemistry. It concerns the transformation of triplet molecular oxygen into a singlet one under photoexcitation in the presence of fullerene C60. Turning off the light returns the system to its previous state with inactive oxygen.
Many spears have been broken in attempts to explain the observed effect without taking into account the open-shell character of the fullerene (see [52,53] and references therein). Until now, the mechanism of the effect has been hidden behind a slogan “triplet state photochemical mechanism” according to a widely accepted scheme [53,54]
The scheme implies the energy transfer from the singlet photoexcited fullerene to the triplet one, which further transfers the energy to convenient triplet oxygen, thus transforming the latter into an active singlet one. The first two stages of this “single-fullerene-molecule” mechanism are quite evident, while the third one, the most important for the final output, is obscure in spite of a number of speculations available [54]. Obviously, this stage efficacy depends on the strength of the IMI between fullerene and oxygen molecules. Numerous quantum chemical calculations show that pairwise interaction in the f–o dyad [C60 + O2] in both singlet and triplet state is practically absent. The UHF computations [55] fully support the previous data and determine the coupling energy of the dyad equal zero in both cases. This poses a serious problem for the explanation of the third stage of the above scheme, forcing the suggestion of the origination of a peculiar IMI between C60 and O2 molecules in the excited state once absent in the ground state.
However, the IMI in the PD solutions is not limited by the fullerene–oxygen interaction only. There are two other interactions, namely: fullerene–fullerene (f–f) and fullerene–solvent (f–s), among which the former is quite significant, thus revealing itself as the fullerene dimerization [56] as well as a considerable amplification of the spectral properties of fullerene solutions [57]. The f–s interaction in the case of aqueous and benzene solutions can be ignored. Besides a significant strength, the f–f interaction possesses some peculiar features caused by the exclusive D-A ability of fullerenes [50]. A significant contribution of the D-A component into the total IMI results in a two-well shape of the potential energy term of a pair of fullerene molecules in the ground state, which is schematically shown in Figure 5a. According to the scheme, the pairwise interaction between the molecules in convenient solutions always leads to the formation of bi-molecular or more complex homoclusters of fullerenes in the vicinity of the R00 minimum on the potential energy curve. The dimerization (as well as oligomerization) is a barrier reaction and does not occur spontaneously. Particular measures should be undertaken to come over the barrier and provide the molecule chemical coupling, in spite of the clusterization is energetically profitable. Photoexcitation is one of the most efficient tools. Therefore, the PD solutions under ambient conditions should involve conglomerates of clustered C60 molecules as shown schematically in Figure 5b, which is experimentally proven in many cases (see for example [57,58,59]). If we remember that =0 in both singlet and triplet state, it becomes clear that oxygen molecules do not interact with either individual fullerene molecule or the molecule clusters, so that the total energy of any f–o dyad [(C60)n-O2] (n= 1, 2, 3….) is just a sum of those related to the dyad components. It is always by 9.93 kcal/mol less in the triplet state due to the difference in the energy of the triplet and singlet oxygen (the UHF energy of and molecules constitutes −27.75 and −17.82 kcal/mol, respectively). Therefore, the ground state of the f–o dyads is triplet.
Computations have shown [56,57] that each pair of fullerene molecules as well as any fullerene cluster of more complex structure formed at the minimum is a charge transfer complex. Their absorption bands related to B2 phototransitions in Figure 5a are located in the UV-visible region. The photoexcitation of either pair or cluster of fullerene molecules within this region produces a pair of molecular ions that quickly relax into the ground state of neutral molecule after the light is switched off. Calculations revealed [55] that, in contrast to neutral C60, both molecular ions C60− and C60+ actively interact with oxygen molecule producing coupling energy and of −10.03 and −10.05 kcal/mol, respectively, referring to molecule and −0.097 and −0.115 kcal/mol in regards to . Therefore, the pristine oxygen molecule is quite strongly held in the vicinity of both molecular ions, thus forming and complexes, as schematically shown in Figure 5c. The complexes are of and compositions of the doublet SpM. Both fullerene ions take the responsibility over the multiplicity, so that two odd electrons of the oxygen molecule “lose their job” and do not more maintain the molecule triplet SpM, thus adding two effectively unpaired electrons to the ND pool of unpaired electrons of the whole complex, as shown in Figure 5e. A dominant contribution of electrons located on oxygen atoms 61 and 62 is clearly seen, thus revealing the most active sites of the complexes. It should be noted that these distributions are intimate characteristics of both complexes, so that not oxygen itself but and complexes as a whole provide the oxidative effect. The effect lasts until the complexes exist and is practically immediately terminated when the complexes disappear when the light is switched off. The obtained results make it possible to suggest the PD mechanism schematically presented in Figure 5d. As shown in the figure, changing SpM from triplet to doublet under photoexcitation due to passing from neutral molecule complex to those based on fullerene molecular ions results in a spin flip in the system of two odd electrons of the oxygen molecule. This approach allows attributing PD effect of fullerene solutions to a specific type of spin-chemical reaction. Particular D-A properties when the molecule can be both the donor and acceptor of electrons are evidently characteristic not only to fullerenes C60 and C70 as well as their derivatives, but to CNT [45] and nanosize graphene molecules [60], thus demonstrating them as an additional characteristic feature of open-shell molecules. This opens a large new branch of a spin photochemistry of the species.
6. Spin Nature of the Reaction Final Products
As follows from the abovementioned, the considered representatives of sp2 nanocarbons enter a chemical reaction equipped with high MCS and a large network of target atoms marked by ACS. The targets are definitely discriminated by the relevant NDA value. Thus, the first step of any additional reactions involving fullerenes C60 will occur with one of the hexagon light blue atoms shown in Figure 1a and Figure 2a. In the case of CNTs and bare graphene molecules, open-end and zigzag-edge atoms play the role, respectively. A lot of information concerning a particular chemical activity of open-end atoms of CNTs and edge atoms of graphene sheets can be found in literature. Actually, convincing evidence that the edge atoms of graphene carry additional electronic (including spin) density was shown by unique experiments on scanning graphene ribbons with atomic resolution [61,62] (see a detailed discussion of the experiments in [14]). As for fullerene C60, a convincing confirmation of the existence of special two rings in this molecule structure was first discovered in 2008 and then confirmed in 2012 in atom-resolved STM [63] and AFM [64] studies. The joint results of the studies are presented on panel I in Figure 6. A scrupulous analysis of the STM image [63] showed that the molecule is attached to the Cu(111) substrate via contact area having a hexagon shape; therewith the area is duplicated
![Carbon 07 00031 g006]() above, thus forming the imaged produced by scanning two hexagon circles of atom located one over the other. In the molecule structure, there is only one pair of such hexagon configurations that coincide with those marked by light blue color in Figure 1a. It is obvious that when the molecule interacts with an extended solid body, the coupling depends on the number of contacts and the interaction strength in each contact. Six light blue carbon atoms best satisfy both requirements, which explains the hexagon configuration of the molecule contact with the flat Cu(111) surface, as seen by STM. The experimental image of AFM scanning (I), in its turn, exhibits homogeneous distribution of the interaction force over the atoms, which is expected if taking into account the equality of NDA values for the atoms (see Figure 2a). A tight connection with exclusive atom-resolved AFM images of sp2 carbon species (see a profound review [65]) and the corresponding NDA distribution is discussed in details in Ref. [14].
above, thus forming the imaged produced by scanning two hexagon circles of atom located one over the other. In the molecule structure, there is only one pair of such hexagon configurations that coincide with those marked by light blue color in Figure 1a. It is obvious that when the molecule interacts with an extended solid body, the coupling depends on the number of contacts and the interaction strength in each contact. Six light blue carbon atoms best satisfy both requirements, which explains the hexagon configuration of the molecule contact with the flat Cu(111) surface, as seen by STM. The experimental image of AFM scanning (I), in its turn, exhibits homogeneous distribution of the interaction force over the atoms, which is expected if taking into account the equality of NDA values for the atoms (see Figure 2a). A tight connection with exclusive atom-resolved AFM images of sp2 carbon species (see a profound review [65]) and the corresponding NDA distribution is discussed in details in Ref. [14].
Figure 6.
(I) STM image of C60 on Cu(111) (left-up-corner insert) that reveals the orientation of C60, while the main image presents the constant-height AFM images of C60 on Cu(111) obtained with a CO tip (see text). (II–VI) Computational spin chemistry of fullerene C60. Monodentant polyderivatives C60H18 (II), C60F18 (III), C60(CN)18 (IV), and C60Cl18 (V) obtained at the 18th step of the successive polyderivatization following NDA algorithm; the same for bidentant poliderivative C60(NH)9 (VI) at the ninth step. See detailed discussion in [44].
Figure 6.
(I) STM image of C60 on Cu(111) (left-up-corner insert) that reveals the orientation of C60, while the main image presents the constant-height AFM images of C60 on Cu(111) obtained with a CO tip (see text). (II–VI) Computational spin chemistry of fullerene C60. Monodentant polyderivatives C60H18 (II), C60F18 (III), C60(CN)18 (IV), and C60Cl18 (V) obtained at the 18th step of the successive polyderivatization following NDA algorithm; the same for bidentant poliderivative C60(NH)9 (VI) at the ninth step. See detailed discussion in [44].

When the first-step derivative is formed, the molecule responds to the action by reconstruction of its {sp2C=C bonds} pool and redistribution of NDA markers. The newly appeared highest NDA mark determines the target carbon atom for the second step and so forth. Therefore, following the highest NDA as descriptor of the target atom at each reaction step determines the NDA algorithm of sp2 carbon molecules polyderivatization. Practical chemistry of sp2 nanocarbons gives many examples confirming the NDA algorithm implementation. The most characteristic results are obtained for fullerene C60 [44]. Exhibited in Figure 6, polyderivatives II–V present a collection of one-dentant C60(X)18 (X=H, F, CN, Cl) species. The first three molecules represent so-called crown structures of local symmetry C3v. These structures were obtained computationally at the 18th step of the successive addition of the addends to C60 following the NDA indication. All the above polyderivatives were synthesized in practice, and their structures identical to shown in Figure 6 were determined (see a detailed discussion of the issue in [44]). If the successive addition of addends in the case of C60H18 (II), C60F18 (III), and C60(CN)18 (IV) proceeds quite similarly, addition of chlorine behaves differently from the first step, due to which the structure of C60Cl8 (V) turns out different. This result correlates with known problems concerning the fullerene chlorination [66]. C60(NH)9 (VI) molecule is a bi-dentant species, and its structure at the ninth step of reaction is of C3v local symmetry, thus showing a similar “crown” configuration, which perfectly fits experimental data.
Another face of the NDA algorithm action can be seen from plottings in Figure 7. Those present the evolution of the heat of formation, ΔH, per-step coupling, Ecpl, and ND quantities in due course of hydrogenation and fluorination of fullerene C60 [67,68]. As seen in the figure, the two reactions are characterized by significantly different energetic parameters with an obvious favoring to fluorination. Actually, the total energy ΔH of hydrides gradually decreases, which favors the polyhydrides formation, until k reaches 15, after which the decreasing temp is slowed down approaching zero at k = 25 and then the energy starts to increase. Since for hydrogen , the Ecpl behavior directly exhibits the above changing in the total energy. The behavior of the total and coupling energies of fluorides is similar, with the only difference concerning much bigger values of both ΔH and Ecpl as well as the absence of the ΔH increasing at k > 25. However, due to a significant negative value of in this case, the coupling energy becomes positive at k >25.
Molecular chemical susceptibility ND is the other characteristic quantifier. As seen in Figure 7b, ND(k) functions are practically identical for both families, gradually decreasing at higher k and approaching zero at k~20–24. Therefore, decreasing Ecpl by absolute value correlates with decreasing MCS ND, or, in other words, with working out the pool of effectively unpaired electrons, which results in a considerable lowering of the reaction activity when k changes from 18–20 to 25–26. According to both characteristics, the reaction is terminated at k > 25. It is important to note that in spite of obvious obstacles, k-high products might be abandoned among the final products. This is due to accumulative character of the reaction until the next addition of the atom pair is energetically favorable. This means that (1) the attachment of a next atom pair will not proceed at positive Ecpl and (2) the accumulation time (and mass yield of the product) will greatly depend on the absolute value Ecpl: the less the value, the longer time is needed. That is why more than four-times difference in the Ecpl absolute values for hydrides in favor of fluorides at k = 18 and their small absolute value result in the termination of hydrogenation process by C60H36 product, while fluorination still continues and is completed by C60F48. The data presented in Figure 7 are in full consent with experimental ones.
Figure 8 briefly summarizes main features concerning polyderivatization of the (5, 5) NGr molecule. The pristine bare molecule is flat and consists of 66 carbon atoms. Chemical portrait of the molecule is given in the upper part of the figure as ACS NDA distribution over atoms. Following NDA algorithm, the formation of framed graphene molecules occurs at the first stage of the molecule chemical modification (see detailed discussion in [31,32,37]), after which the basal plane becomes the battlefield. Final products are produced in the course of topochemical reaction due to dependence on such important factors as (1) fixation or free standing of the molecule, (2) accessibility of either one or both sides of the molecule plane, and (3) chemical addends in use. The role of these three factors can be seen in Figure 8, where a collection of (5, 5) NGr polyderivatives related to the maximum covering of the parent molecule by addends is exhibited. Thus, the left column presents graphene hydrides obtained computationally in the course of successive hydrogenation of the parent molecule following NDA algorithm at different conditions. As seen, their structures differ drastically, changing from regular graphane one (a) to highly bent pancake (c) [69]. Not shown in the figure, a canape-like structure of hydride obtained under one-side access of a fixed molecule completes a full list of the products. Experimentally, regular graphane and amorphous canape-like structures of hydrogenated graphene membranes were observed under the relevant conditions [70]. When hydrogen is substituted by fluorine (see Figure 8a), the structure of polyfluorides changes drastically, supporting, in general, the (a)–(c) tendency of hydrides. However, none of the regular graphane-like structures were obtained. A thorough experimental study of graphene fluorides [71] confirms this conclusion. The situation becomes still more cumbersome when hydrogen is substituted with hydroxyls (Figure 8b) or a combination of oxygen atoms and hydroxyls (Figure 8c). The last two cases present selected variants of numerous configurations related to graphene molecule oxygenation [72].
Therefore, polyderivatization is extremely complicated and covers a great number of final products. As mentioned earlier, addition of each addend at every step is accompanied by immediate reconstruction of the {sp2C=C bonds} pool. The pool is highly labile and readily responds to any perturbation. This NDA-reconstructing distribution makes the polyderivatives production highly variable due to high sensitivity to any current situation, thus depriving such names as “graphene hydride”, “graphene oxide”, “graphene fluoride”, and so forth of an exact chemical formulae and allowing speaking about large classes of substances that are related to the above names. Therefore, the chemistry of graphene molecules is not a chemistry of a molecule, but the chemistry of a molecular class.
So far, we have dealt with products of the highest complete derivatization. The mass-produced “graphene oxide” (GO) class is their best-known representative [73]. However, there are still innumerable products that are not completely derivated. Framed graphene molecules are of such species. These molecules became a regular participant in the scientific agenda a decade ago when a new high-tech material, reduced graphene oxide (rGO), entered practical graphenics. The product is not synthesized from the bottom, but is produced from the top using GO as a parent species. Evidently, a large GO class gives life to a new rGO class, additionally subjected to variations due to variable conditions of the reduction procedures (see a profound review [74]). rGO molecules present fragments of the honeycomb domains framed by different atomic groups, depending on the reduction technology. The mass product presents a powder of amorphous structure consisting of layered stacks of a few-nanometer thickness and from first nanometers to submicron dimension in the lateral direction. Interlayer distance is close to that of graphite, thus justifying a flat graphene-like structure of the layers (see as example, a neutron scattering study of different-origin rGOs [75]). Accordingly, in the literature, you can often find a reference to rGO as graphene, due to which it was suggested to cite it as “technical graphene” [76].
A long time from the beginning of the graphene era, there was an opinion that chemical derivatives of graphene, mainly GO, rGO, and teflon, came into life together with graphene itself. The first breakdown was caused by discovering that natural amorphous carbon, known as shungite carbon, is a natural deposit containing a million tons of rGO [77]. Then, similar conclusions were made about the natural anthraxolite [78] and anthracite [79]. In time, it came to the industrial multi-tonnage carbon blacks as well [79,80,81]. Therefore, having been known for more than one-and-a-half thousand years, amorphous carbon is undergoing a rebirth and appears as agglomerative compositions of framed graphene molecules, thus becoming a special subject of modern nanotechnology. A recent extended study was devoted to a detailed investigation of the structure and chemical composition of amorphous carbons of the highest carbonization [79,80,81,82]. For the first time, reasonable models of graphene molecules, which are the basic structural units (BSUs) of the studied solids and which correspond to both their structure and chemical composition, including a definite C:O:H atom relation, were suggested. A set of selected models is shown in Figure 9. The same (5, 5) NGr molecule, which is commensurate with experimentally determined BSUs of shungite carbon, anthraxolite, and carbon black 632, was chosen as parent structure in all the cases. The choice allows drawing the main attention to the framing areas of the models without distracting from the structure of the graphene domains. As seen in the figure, BSUs of amorphous carbons belong to the class of graphene oxyhydrides, differing by the hydrogen content and chemical composition of oxygen containing groups. Bright spots on the NDA distribution maps reveal the radical character of the species. Later on, the models were corrected based on IR reflection [81] and Raman scattering [82] data. The correction concerned the structure-chemical pattern of the heteroatom distribution around the pristine (5, 5) NGr molecule, keeping the relation of C:O:H components fixed and the radical character of the models conserved.
Revealing the molecular structure of amorphous carbon significantly changes our view of the world. We live on a land containing a high amount of amorphous carbon, we breathe air with a high content of combustion products of organic substances, we eat food as polluted as air, and we drink water extracted from underground sources or rivers suffering from carbon pollution. Is it not time to ask ourselves if we realize that in most cases we are dealing with radicalized graphene molecules and that this circumstance casts doubt on our safety? Obviously, the answers concern the post-reaction existence of the radical molecules. Let us examine which answers we can get now.
7. Post-Reaction Storage of the Spin Chemistry Products
UHF calculations reveal that all the models shown in Figure 9 are radicals. Characterizing by ND value, the molecules form a following series of data: 27.4 e (shungite carbon), 22.2 e (anthraxolite), 29.7 e (carbon black 632), and 30.9 e (carbon black 624) [81]. The quantities are quite big, evidencing strong radicalization. As seen in Figure 9, black balls of the NDA portraits in the circumference areas of the molecules clearly exhibit a fully inhibited chemical ability of both heteroatoms and carbon atoms to which the former are attached. Nevertheless, the remaining part of the molecules’ circumferences still remains highly active and easily accessible to not only gaseous reagents, but to bulky ones as well, even once aggravated with sterical constrains in the latter case.
Due to high radicalization of each suggested model and since the models, in general, are fully consistent with the discussed structural–compositional characteristics of the studied ACs [81], the authors made an astonishing conclusion that sp2 amorphous carbons are conglomerates of stable radicals. In itself, this conclusion, however unexpected it may be, is not knockdown. Currently, stable organic radicals in the form of stable tetrabenzo-Chichibabin’s hydrocarbons [83], materials based on long acenes [84,85], radical polymers [86], and synthetic metals [87] occupy an increasing part of the agenda of modern chemistry. Graphene fragments were not left aside as well, as can be judged by a series of in-depth reviews of the case [88,89,90,91,92,93,94]. However, it is difficult to imagine that the huge amount of sp2 amorphous carbons, both natural and engineered, as well as their exclusively broad involvement in various chemical and technical processes for more than a thousand years could not disregard such an important characteristic of the substance if it existed. Nevertheless, in fact, by now there have not been communications joining radical terminology and the vast available data concerning sp2 amorphous carbons. At the same time, specific peculiarities concerning metal free carbocatalysis [95,96,97,98,99,100,101,102,103,104,105,106] (see a brief review of the latter state of art [107]) and electron-spin resonance [108,109,110,111,112,113,114,115] have been really observed. However, until now, they have not been connected with specific radical properties of sp2 carbons as well. Against this background, a study [116] that voiced the term “radical” in relation to sp2 amorphics for the first time when attempting to explain carbonaceous soot inception and growth in term of resonance-stabilized hydrocarbonradical chain reactions is quite revolutionary.
Restoration of BSUs radicals in their rights confronts us with new problems to be solved, and the first one is to understand why sp2 carbon molecular radicals are stable. The appeal should be attributed to fullerenes and CNTs as well, the radical essence of which has been convincingly established [44]. The question of the stabilization of open-shell molecules has been raised repeatedly (see reviews [23,24,89,93]). However, the time-dependent stability of molecules was determined not directly, but by a qualitative comparison of calculated thermodynamic factors [117]. The procedure was applied to molecules with terminated DBs, such as PAHs or periacenes. For these molecules, it was established that spin-delocalized character of the molecule radicalization, provided with the conjugation of sp2 electrons over the total number of carbon atoms, definitely favors the stabilization. Additionally, nearly degenerated spin–triplet gap EST as well as incorporation of heteroatoms (O, N, S) inside benzenoid units or outside the latter support the stabilization as well. All these factors are typical for graphene molecules, both bare and framed, such as, say, (5, 5) NGr and peripentacene in Figure 5 as well as semi-bare or/and semi-framed BSU molecules in Figure 9. The latter, as we know exactly, do live for a long time [79].
Another problem of these bodies concerns the real implementation of BSU structures from the available variety of possible models. The model’s total energies Egr evidence a potential ability only, while the nature of empirical observations concern the chemical kinetics rather than the thermodynamic stability of the products. Reaction occurrence is governed by parameters that control the interrelation between reactants and products, forming free energy basins separated by barriers of different high. One can easily imagine how complicated the picture of the basins is when one of the reactant is multi-target. Until now, a possibility to quantitatively consider the relevant problems related to the formation of, say, shungite carbon BSUs, has seemed impossible. However, the appearance of a new method, called by its authors “a multi class harmonic linear discriminant analysis” (MC-HLDA) [118], presenting metadynamics with discriminants as a tool for understanding chemistry, inspires some optimism that in the near future, similar complex problems can be solved.
The idea of a particular role of kinetics in the case of multi-target chemical compounds with highly delocalized spin density puts the time in the avant-garde row of particularly significant parameters. The chemistry of sp2 nanocarbons requires the determination of the time of life of manufactured products. Despite the revolutionary contribution of sp2 nanocarbons to modern chemistry, this area itself is still young, so the question of lifetime may seem premature. Nevertheless, evidence of the legitimacy of such an issue has already been obtained in synthetic chemistry. Thus, it was found that the lifetime of the simplest sp2 radicals, which are well-known polyacenes, is different. Long-lived members of this series are naphthalene, anthracene, tetracene, and pentacene. As for longer acenes, the lifetime drastically shortens when the number of BUs increases, so that hexacene has been still observed for a short time, while the higher species cannot even be recorded as a result of chemical synthesis [24,119,120]. Synthesis of multinuclear PAHs imitating graphene molecules by Müllen’s team [121] also faces the problem of sustainability of the final products. The undoubted success of the synthesis is the existence of stable graphene materials, such as mass-produced GO, rGO, and graphene quantum dots based on it. Although these materials have not been even two decades old and their temporal stability has not yet been truly investigated, the time-instability of GO has been rigidly fixed [75]; many chemists are familiar with spontaneous oxidation of fullerenes.
Besides synthetic laboratories, there is a unique natural laboratory, for which carbon is an absolute favorite. The laboratory operates in a time scale of billions (shungite carbon) and millions (anthraxolite, anthracite) of years. The time is large enough to expect that all chemical transformations, which could have happened with sp2 carbons, already occurred. Consequently, compounds exist in size, shape, and chemical composition, which no longer change. A careful analysis of these properties allows us to highlight a number of unique characteristics of natural amorphous carbons. (1) The average BSUs size of shungite carbon, anthraxolite, and anthracite is of 1.5 nm, nondependent on the local place of deposits [75,79]. (2) The chemical composition of these mineral BSUs is similar [79]. Apparently, these factors are favorable for the radical stabilization. At the same time, the BSUs preserve their radical character, which can be opened by changing the conditions of existence and/or storage of the substances. The latter lays the foundation of particular features of shungite carbon, favoring its application in medical treatment [122], biology [123], non-linear optics [124], mechanics [125], and so on [126]. So far, anthracite and anthraxolite have not been known from this side, while many exciting discoveries can be expected.
It becomes obvious as well that the properties of shungite carbon itself and in the vicinity to other minerals should be different due to a possible activation of the radical ability of the former. Actually, any mineral nucleus is an atomic cluster covered with DBs, thus having a radical nature. The latter are saturated when stacking with other nuclei promotes the mineral growth. Obviously, the termination of the bonds by another’s addends terminates the growth [38]. Let us demonstrate the said on the example of the growth of a particular quartz in the presence of shungite carbon. (SiO4)48 cluster in Figure 10a presents a silica nucleus, leading to the growth of α-quartz. The cluster is composed in such a way that the consequent growth can occur from the basal top plane of the cluster that accommodates nine silicon atoms with one DB each. The shungite carbon BSU molecule (Figure 10b), which appears nearby, reacts with the cluster almost barrierlessly and is bound to it by two Si–C bonds, irrespective of the initial arrangement of the reactants (Figure 10e,f). The coupling energy constitutes −139.93 and −124.35 kcal/mol, respectively. Evidently, the presence of the molecule prevents from joining two silica clusters for the quartz growth to be continued. When the carbon molecule number is big, they together may form a “patchwork fabric” to cover or envelope the silica nucleus. Assuming the nucleus spherical shape, geometry allows the evaluation of the sphere radius R, when it is fully covered by the fabric consisting of ~1.5 × 1.5 nm2 patches, which gives the value of R~80–100 nm. Therefore, it is possible to expect the formation of a mineral matter consisting of peculiar “chocolate candies” with a hard silica core inside, surrounded by shungite–carbon shell.
The problem under discussion did not arise from scratch. It was stimulated by a thorough physicochemical study of conditional shungite rocks with low carbon content [127]. Such a component is always present in shungite deposits. As occurred, the mineral matter actually consists of agglomerates of close-to-spherical nanoparticles presenting a crystalline inner core of perfect α-quartz surrounded by shungite–carbon shell. The size of quartz core is of ~60–80 nm irrespectively of the deposits place. The shell can be easily separated from the core. The study of this carbon mass has revealed its full similarity to that one related to high-carbon shungite rocks. It is evident that this first observation of a particular symbiosis of shungite carbon and silica should not be the only one. It can be assumed that the discovered phenomenon, which is self consistently explained from the standpoint of spin chemistry of nanocarbons, opens a new page in the history of geology, devoting it to the spin geochemistry.
8. Conclusions
The paper presents a self-consistent overview of a selected number of the top issues related to sp2 nanocarbons from the viewpoint of an experienced long-working user of computational quantum chemistry. sp2 Nanocarbons are considered to be specific objects, the electronic structure features of which are presented in light of the general concept of emergent chemical phenomena. The latter are a consequence of a quantum phase transition caused by the spin symmetry breaking due to the correlation of pz odd electrons. This correlation depends on the distance between the odd electrons and becomes noticeable when the shortest one, defined by C=C bond length, exceeds critical value . The UHF formalism clearly evidences the broken symmetry occurrence and perfectly suits self-consistent description of the issue. The UHF emergents, such as (i) open-shell character of electron spin-orbitals; (ii) splitting and/or spin polarization of electron spectrum; (iii) spin-mixed character of the ground state and, consequently, depriving the exact spin multiplicity of electronic states; as well as (iv) a pool of local spins at zero total spin density, are empirically supported and convincingly certified. They significantly expand the concept of the ground state of sp2 nanocarbons and give a clear vision of the spin features of their chemistry as the chemistry of stable radicals. Once multitarget and spatially extended, stable radicals of sp2 nanocarbons should be considered from the standpoint of spin-delocalized topochemistry. The latter is based on both spin density and chemical activity delocalization over molecules’ atoms, on the one hand, and on selectively pronounced barriers of different reactions exhibiting topological essence of intermolecular interaction, on the other. Accordingly, sp2 carbon radicals reveal a peculiar topokinetics that lay the foundation of their stability. The stability is time-marked, which entails the emergence of a new characteristic term in the modern chemistry of sp2 carbons—the species lifetime. A number of examples exhibiting main traits of computational spin chemistry of fullerene C60, a fragment of (4, 4) SWCNT, and a family of graphene molecules on the basis of a parent (5, 5) NGr one are presented. A perfect fitting of calculated and experimental data is discussed. Particular attention is given to graphene molecules presenting basic structure units of synthetic and natural amorphous carbons. The stable-radical character of the latter allows raising questions about spin geochemistry due to the ubiquitous presence of amorphous carbon in nature.
Funding
This research received no external funding.
Acknowledgments
The author much appreciates the fruitful discussions with E. Brändas, J. Karwowski, O. Ori, E. Orlenko, I. Natkaniec, Ye. Golubev, and N. Rozhkova. This publication has been supported by the RUDN University Strategic Academic Leadership Program.
Conflicts of Interest
The author declare no conflict of interest.
Abbreviations
| ACS | atomic chemical susceptibility |
| BSS | broken spin symmetry |
| BSU | basic structural unit |
| BU | benzenoid unit |
| CI | configuration interaction |
| CNT | carbon nanotube |
| DBs | dangling bonds |
| DFT | density functional theory |
| DMRG | density matrix renormalization group |
| GO | graphene oxide |
| IMI | intermolecular interaction |
| MCS | molecular chemical susceptibility |
| NDA | effectively unpaired electrons fraction at atom A |
| ND | total number of effectively unpaired electrons |
| PAH | polyaromatic hydrocarbons |
| PH | photodynamics |
| RAS-SF | restricted active space spin–flip |
| rGO | reduced graphene oxide |
| RHF | restricted Hartree-Fock |
| SC | spin contamination |
| {sp2C=C bonds} | a complete set o C=C bonds |
| SpDA | spin density fraction at atom A |
| SpDtot | total spin density |
| SpM | spin multiplicity |
| SWCNT | single walled carbon nanotube |
| UDFT | unrestricted density functional theory |
| UHF | unrestricted Hartree-Fock |
References
- Pople, J.A.; Nesbet, R.K. Self-Consistent Orbitals for Radicals. J. Chem. Phys. 1954, 22, 571. [Google Scholar] [CrossRef]
- Löwdin, P.-O. Quantum Theory of Many-Particle Systems. III. Extension of the Hartree-Fock Scheme to Include Degenerate Systems and Correlation Effects. Phys. Rev. 1955, 97, 1509. [Google Scholar] [CrossRef]
- Löwdin, P.-O. Correlation Problem in Many-Electron Quantum Mechanics I. Review of Different Approaches and Discussion of Some Current Ideas. Adv. Chem. Phys. 1958, 2, 209. [Google Scholar] [CrossRef]
- Ray, S.S.; Manna, S.; Ghosh, A.; Chaudhuri, R.K.; Chattopadhyay, S. Multireference Perturbation Theory with Improved Virtual Orbitals for Radicals: More Degeneracies, More Problems. Int. J. Quant. Chem. 2018, 119, e25776. [Google Scholar] [CrossRef]
- Takatsuka, K.; Fueno, T.; Yamaguchi, K. Distribution of Odd Electrons in Ground-State Molecules. Theor. Chim. Acta 1978, 48, 175. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Davidson, E.R. Distribution of Effectively Unpaired Electrons. Chem. Phys. Lett. 2000, 330, 161. [Google Scholar] [CrossRef]
- Anderson, P.W. More Is Different. Science 1972, 177, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laughlin, R.B. Nobel Lecture: Fractional Quantization. Rev. Mod. Phys. 1999, 71, 863. [Google Scholar] [CrossRef] [Green Version]
- Laughlin, R.B.; Pines, D. The Theory of Everything. Proc. Natl. Acad. Sci. USA 2000, 97, 28. [Google Scholar] [CrossRef] [Green Version]
- Yannouleas, C.; Landman, U. Symmetry Breaking and Quantum Correlations in Finite Systems: Studies of Quantum Dots and Ultracold Bose Gases and Related Nuclear and Chemical Methods. Rep. Prog. Phys. 2007, 70, 2067. [Google Scholar] [CrossRef] [Green Version]
- Pavarini, E.; Koch, E.; Schollwöck, U. (Eds.) Emergent Phenomena in Correlated Matter, Autumn School, Jülich, 23–27 September 2013; Forschungszentrum Jülich Zentralbibliothek: Jülich, Germany, 2013. [Google Scholar]
- Putz, M.V.; Ori, O.; Diudea, M.V.; Zefler, B.; Pop, R. Distance, Symmetry, and Topology in Carbon Nanomaterials; (Carbon Materials: Chemistry and Physics); Ashrafi, A.R., Diudea, M.V., Eds.; Springer: Berlin, Germany, 2016; Volume 9, p. 345. [Google Scholar]
- Sheka, E.F.; Popova, N.A.; Popova, V.A. Physics and Chemistry of Graphene. Emergentness, Magnetism, Mechanophysics and Mechanochemistry. Phys. Uspekhi 2018, 61, 645. [Google Scholar] [CrossRef]
- Fucutome, H. Unrestricted Hartree–Fock Theory and Its Applications to Molecules and Chemical Reactions. Int. J. Quant. Chem. 1981, 20, 955. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Saito, T.; Nakanishi, Y.; Kataoka, Y.; Matsui, T.; Kawakami, T.; Okumura, M.; Yamaguchi, K.J. Spin Contamination Error in Optimized Geometry of Singlet Carbene (1A1) by Broken-Symmetry Method. Phys. Chem. A 2009, 113, 15041. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Bulik, I.W.; Jiménez-Hoyos, C.A.; Henderson, T.M.; Scuseria, G.E. Proper and Improper Zero Energy Modes in Hartree-Fock Theory and Their Relevance for Symmetry Breaking and Restoration. J. Chem. Phys. 2013, 139, 154107. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Davidson, E.R.; Yang, W. Nature of Ground and Electronic Excited States of Higher Acenes. Proc. Natl. Acad. Sci. USA 2016, 113, E5098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hachmann, J.; Dorando, J.J.; Aviles, M.; Chan, G.K.-L. The Radical Character of the Acenes: A Density Matrix Renormalization Group Study. J. Chem. Phys. 2007, 127, 134309. [Google Scholar] [CrossRef] [Green Version]
- Casanova, D.; Head-Gordon, M. Restricted Active Space Spin-Flip Configuration Interaction Approach: Theory, Implementation and Examples. Phys. Chem. Chem. Phys. 2009, 11, 9779. [Google Scholar] [CrossRef]
- Jacob, C.R.; Reiher, M. Spin in Density-Functional Theory. Int. J. Quantum Chem. 2012, 112, 3661. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, I.G. Problems in DFT with the Total Spin and Degenerate States. Int. J. Quant. Chem. 2007, 107, 2595. [Google Scholar] [CrossRef]
- Kaplan, I.G. Symmetry Properties of the Electron Density and Following From It Limits on the KS-DFT Applications. Mol. Phys. 2018, 116, 658. [Google Scholar] [CrossRef]
- Shee, J.; Arthur, E.J.; Zhang, S.; Reichman, D.R.; Friesner, R.A.J. Singlet–Triplet Energy Gaps of Organic Biradicals and Polyacenes with Auxiliary-Field Quantum Monte Carlo. Chem. Theory Comput. 2019, 15, 4924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishna, T.Y.; Zeng, W.; Lu, X.; Wu, J. From Open-Shell Singlet Diradicaloids to Polyradicaloids. Chem. Comm. 2018, 54, 2186. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C.; Bonaccorso, F.; Fal’Ko, V.; Novoselov, K.S.; Roche, S.; Bøggild, P.; Borini, S.; Koppens, F.H.; Palermo, V.; Pugno, N.; et al. Science and Technology Roadmap for Graphene, Related Two-Dimensional Crystals, and Hybrid Systems. Nanoscale 2015, 7, 4598–4810. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.S.; Fal’ko, V.I.; Colombo, L.; Gellert, P.R.; Schwab, M.G.; Kim, K. A Roadmap for Grapheme. Nature 2012, 490, 192. [Google Scholar] [CrossRef]
- Kauling, A.P.; Seefeldt, A.T.; Pisoni, D.P.; Pradeep, R.C.; Bentini, R.; Oliveira, R.V.B.; Novoselov, K.S.; Castro Neto, A.H. The Worldwide Graphene Flake Production. Adv. Mater. 2018, 30, 1803784. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Salinas, M.E.; Carreras, A.; Casanova, D. Triangular Graphene Nanofragments: Open-Shell Character And Doping. Phys. Chem. Chem. Phys. 2019, 21, 9069. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E.F.; Chernozatonskii, L.A. Chemical Reactivity and Magnetism of Graphene. Int. J. Quant. Chem. 2010, 110, 1938. [Google Scholar] [CrossRef] [Green Version]
- Sheka, E.F. Nanocarbons through Computations: Fullerenes, Nanotubes, and Graphene. In Nanoscience and Nanotechnologies; Kharkin, V., Bai, C., Awadelkarim, O.O., Kapitaza, S., Eds.; Eolss Publishers: Abu Dhabi, UAE, 2011. [Google Scholar]
- Sheka, E.F. Computational Strategy for Graphene: Insight from Odd Electrons Correlation. Int. J. Quant. Chem. 2012, 112, 3076. [Google Scholar] [CrossRef] [Green Version]
- Sheka, E.F. Molecular Theory of Graphene. In Advances in Quantum Methods and Applications in Chemistry, Physics, and Biology; Hotokka, M., Brändas, E., Maruani, J., Eds.; Spinger: Berlin, Germany, 2013; p. 249. [Google Scholar]
- Sheka, E.F. The Uniqueness of Physical and Chemical Natures of Graphene: Their Coherence and Conflicts. Int. J. Quantum Chem. 2014, 114, 1079. [Google Scholar] [CrossRef] [Green Version]
- Sheka, E.F. Spin Effects of sp2 Nanocarbons in Light of Unrestricted Hartree-Fock Approach and Spin-Orbit Coupling Theory. In Quantum Systems in Physics, Chemistry, and Biology: Advances in Concepts and Applications; Tadjer, A., Pavlov, R., Maruani, J., Brändas, E., Delgado-Barrio, G., Eds.; Springer: Berlin, Germany, 2017; p. 39. [Google Scholar]
- Budyka, M.F.; Sheka, E.F.; Popova, N.A. Graphene Quantum Dots: Theory and Experiment. Rev. Adv. Mat. Sci. 2017, 51, 35. [Google Scholar]
- Sheka, E.F. Dirac Material Graphene. Rev. Adv. Mat. Sci. 2018, 53, 1. [Google Scholar] [CrossRef] [Green Version]
- Sheka, E.F. Spin Chemical Physics of Graphene; Pan Stanford: Singapore, 2018. [Google Scholar]
- Hoffmann, R. Small but Strong Lessons from Chemistry for Nanoscience. Ang. Chem. Int. Ed. 2013, 52, 93. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E.F.; Razbirin, B.S.; Nelson, D.K. Continuous Symmetry of C60 Fullerene and Its Derivatives. J. Phys. Chem. A 2011, 115, 3480. [Google Scholar] [CrossRef] [Green Version]
- Sheka, E.F.; Popova, V.A. Virtual Spectrometer for Radicals Vibrations. 1. Polyacenes and Fullerenes. arXiv 2020, arXiv:2008.08645. [Google Scholar]
- Sheka, E.F. Stretching and Breaking of Chemical Bonds, Correlation of Electrons, and Radical Properties of Covalent Species. Adv. Quant. Chem. 2015, 70, 111. [Google Scholar]
- Zayets, V.A. CLUSTER-Z1: Quantum-Chemical Software for Calculations in the s,p-Basis; Institute of Surface Chemistry, National Academy of Sciences of Ukraine: Kiev, Ukraine, 1990. (In Russian) [Google Scholar]
- Zabrodsky, H.; Peleg, S.; Avnir, D.J. Continuous Symmetry Measures. Am. Chem. Soc. 1992, 114, 7843. [Google Scholar] [CrossRef]
- Sheka, E.F. Fullerenes: Nanochemistry, Nanomagnetism, Nanomedicine, Nanophotonics; T&F CRC Press: London, UK, 2011. [Google Scholar]
- Sheka, E.F.; Chernozatonsky, L.A. Broken Symmetry Approach and Chemical Susceptibility of Carbon Nanotubes. Int. J. Quant. Chem. 2010, 110, 1466. [Google Scholar] [CrossRef] [Green Version]
- Sheka, E.F.; Orlenko, E.V. Spin-Orbit Coupling of sp2 Nanocarbons and Magnetism of Fullerene C60 in View of Spin Peculiarities of Unrestricted Hartree-Fock Solution. Fuller. Nanotub. Carbon Nanostruct. 2017, 25, 289. [Google Scholar] [CrossRef]
- Sheka, E.F. “Chemical portrait” of Fullerene Molecules. J. Str. Chem. 2006, 47, 593. [Google Scholar] [CrossRef] [Green Version]
- Hauke, F.; Hirsch, A. Carbon Nanotubes and Related Structures Synthesis, Characterization, Functionalization, and Applications; Guldi, D.M., Martín, N., Eds.; Wiley-VCH: Weinheim, Germany, 2010; p. 135. [Google Scholar]
- Sheka, E.F. Topochemistry of Spatially Extended sp2 Nanocarbons. Fullerenes, Nanotubes, and Graphene. In Topological Modelling of Nanostructures and Extended Systems. Carbon Materials: Chemistry and Physics; Ashrafi, A.R., Cataldo, F., Iranmanesh, A., Ori, O., Eds.; Springer: Berlin, Germany, 2013; Volume 7, p. 137. [Google Scholar]
- Sheka, E.F. Intermolecular Interaction in C60-Based Electron Donor–Acceptor Complexes. Int. J. Quant. Chem. 2004, 100, 388. [Google Scholar] [CrossRef]
- Kasermann, F.; Kempf, C. Buckminsterfullerene and Photodynamic Inactivation of Viruses. Rev. Med. Virol. 1998, 8, 143. [Google Scholar] [CrossRef]
- Piotrovski, L.B. Carbon Nanotechnology; Dai, L., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; p. 235. [Google Scholar]
- Da Ros, T. Twenty Years of Promises: Fullerene in Medicinal Chemistry. In Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes; Cataldo, F., Da Ros, T., Eds.; Springer: Berlin, Germany, 2008; p. 1. [Google Scholar]
- Mroz, P.; Tegos, G.P.; Gali, H.; Wharton, T.; Sarna, T.; Hamblin, M.R. Fullerenes as Photosensitizers in Photodynamic Therapy. In Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes; Cataldo, F., Da Ros, T., Eds.; Springer: Berlin, Germany, 2008; p. 79. [Google Scholar]
- Sheka, E.F. Nanophotonics of Fullerene 1. Chemistry and Medicine. Nanosci. Nanothech. Let. 2011, 3, 28. [Google Scholar] [CrossRef]
- Sheka, E.F. Donor-Acceptor Interaction and Fullerene C60 Dimerization. Chem. Phys. Lett. 2007, 438, 119. [Google Scholar] [CrossRef]
- Sheka, E.F.; Razbirin, B.S.; Starukhin, A.N.; Nelson, D.K.; Degunov, M.Y.; Troshin, P.A.; Lyubovskaya, R.N. Fullerene-Cluster Amplifiers and Nanophotonics of Fullerene Solutions. J. Nanophot. 2009, 3, 033501. [Google Scholar] [CrossRef]
- Nath, S.; Pal, H.; Palit, D.K.; Sapre, A.V.; Mitta, J.P. Aggregation of Fullerene, C60, in Benzonitrile. J. Phys. Chem. B 1998, 102, 10158. [Google Scholar] [CrossRef]
- Samal, S.; Geckeler, K.E. Unexpected Solute Aggregation in Water on Dilution. J. Chem. Soc. Chem. Commun. 2001, 2224. [Google Scholar] [CrossRef] [PubMed]
- Razbirin, B.S.; Rozhkova, N.N.; Sheka, E.F.; Nelson, D.K.; Starukhin, A.N. Fractals of Graphene Quantum Dots in Photoluminescence of Shungite. J. Exp. Theor. Phys. 2014, 118, 735. [Google Scholar] [CrossRef] [Green Version]
- van der Lit, J.; Boneschanscher, M.P.; Vanmaekelbergh, D.; Ijäs, M.; Uppstu, A.; Ervasti, M.; Harju, A.; Liljeroth, P.; Swart, I. Suppression of Electron–Vibron Coupling in Graphene Nanoribbons Contacted via a Single Atom. Nat. Commun. 2013, 4, 2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, J.H.; Lin, Y.C.; He, K.; Koshino, M.; Suenaga, K. Atomic Level Spatial Variations of Energy States along Graphene Edges. Nano Lett. 2014, 14, 6155. [Google Scholar] [CrossRef]
- Larsson, K.J.A.; Elliott, S.D.; Greer, J.C.; Repp, J.; Meyer, G.; Allenspach, R. Orientation of Individual C60 Molecules Adsorbed on Cu(111): Low-Temperature Scanning Tunneling Microscopy and Density Functional Calculations. Phys. Rev. B 2008, 77, 115434. [Google Scholar] [CrossRef] [Green Version]
- Gross, L.; Mohn, F.; Moll, N.; Schuler, B.; Criado, A.; Guiti, E.; Peña, D.; Gourdon, A.; Meyer, G. Bond-Order Discrimination by Atomic Force Microscopy. Science 2012, 337, 1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, L.; Schuler, B.; Pavliček, N.; Fatayer, S.; Majzik, Z.; Moll, N.; Peña, D.; Meyer, G. Atomic Force Microscopy for Molecular Structure Elucidation. Angew. Chem. Int. Ed. 2018, 57, 3888. [Google Scholar] [CrossRef] [PubMed]
- Troshin, P.A.; Troshina, O.A.; Lyubovskaya, R.N.; Razumov, V.F. Functional Derivatives of Fullerenes. In Synthesis and Applications to Organic Electronics and Biomedicine; Ivanovo State University: Ivanovo, Russia, 2009. (In Russian) [Google Scholar]
- Sheka, E.F. Computational Synthesis of Hydrogenated Fullerenes from C60 to C60H60. J. Mol. Mod. 2011, 17, 1973. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E.F. Step-Wise Computational Synthesis of Fullerene C60 derivatives. Fluorinated Fullerenes C60F2k. J. Exp. Theor. Phys. 2010, 111, 395. [Google Scholar]
- Sheka, E.F.; Popova, N.A. Odd-Electron Molecular Theory of Graphene Hydrogenation. J. Mol. Mod. 2012, 18, 3751. [Google Scholar] [CrossRef]
- Elias, D.C.; Nair, R.R.; Mohiuddin, T.M.G.; Morozov, S.V.; Blake, P.; Halsall, M.P.; Ferrari, A.C.; Boukhvalov, D.W.; Katsnelson, M.I.; Geim, A.K.; et al. Control of Graphene’s Properties by Reversible Hydrogenation: Evidence for Graphane. Science 2009, 323, 610. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.R.; Ren, W.; Jalil, R.; Riaz, I.; Kravets, V.G.; Britnell, L.; Blake, P.; Schedin, F.; Mayorov, A.S.; Yuan, S.; et al. Fluorographene: A Two-Dimensional Counterpart of Teflon. Small 2010, 6, 2877. [Google Scholar] [CrossRef]
- Sheka, E.F.; Popova, N.A. Molecular Theory of Graphene Oxide. Phys. Chem. Chem. Phys. 2013, 15, 13304. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liu, L.; Li, F. Graphene Oxide: Physics and Applications; Springer: Berlin, Germany, 2015. [Google Scholar]
- Eng, A.Y.C.; Chua, C.K.; Pumera, M. Refinements to the Structure of Graphite Oxide: Absolute Quantification of Functional Groups via Selective Labelling. Nanoscale 2015, 7, 20256. [Google Scholar] [CrossRef]
- Natkaniec, I.; Sheka, E.F.; Drużbicki, K.; Hołderna-Natkaniec, K.; Gubin, S.P.; Buslaeva, E.Y.; Tkachev, S.V. Computationally Supported Neutron Scattering Study of Parent and Chemically Reduced Graphene Oxide. J. Phys. Chem. C 2015, 119, 18650. [Google Scholar] [CrossRef]
- Sheka, E.F.; Golubev, E.A. Technical Graphene (Reduced Graphene Oxide) and Its Natural Analog (Shungite). Tech. Phys. 2016, 61, 1032. [Google Scholar] [CrossRef]
- Sheka, E.F.; Rozhkova, N.N. Shungite as the Natural Pantry of Nanoscale Reduced Graphene Oxide. Int. J. Smart. Nanomat. 2014, 5, 1. [Google Scholar] [CrossRef]
- Golubev, Y.A.; Isaenko, S.I.; Prikhodko, A.S.; Borgardt, N.I.; Suvorova, E.I. Raman Spectroscopic Study of Natural Nanostructured Carbon Materials: Shungite vs. Anthraxolite. Eur. J. Mineral. 2016, 28, 545. [Google Scholar] [CrossRef]
- Golubev, Y.A.; Rozhkova, N.N.; Kabachkov, E.N.; Shul’ga, Y.M.; Natkaniec-Hołderna, K.; Natkaniec, I.; Antonets, I.V.; Makeev, B.A.; Popova, N.A.; Popova, V.A.; et al. sp2 Amorphous Carbons in View of Multianalytical Consideration: Normal, Expected and New. J. Non-Cryst. Sol. 2019, 524, 119608. [Google Scholar] [CrossRef] [Green Version]
- Sheka, E.F.; Hołderna-Natkaniec, K.; Natkaniec, I.; Krawczyk, J.X.; Golubev, Y.A.; Rozhkova, N.N.; Kim, V.V.; Popova, N.A.; Popova, V.A. Computationally Supported Neutron Scattering Study of Natural and Synthetic Amorphous Carbons. J. Phys. Chem. C 2019, 123, 15841. [Google Scholar] [CrossRef]
- Sheka, E.F.; Natkaniec, I.; Ipatova, E.U.; Golubev, Y.A.; Kabachkov, E.N.; Popova, V.A. Heteroatom Necklaces of sp2 Amorphous Carbons. XPS Supported INS and DRIFT Spectroscopy. Fuller. Nanotub. Carbon Nanostruct. 2020, 28, 1010. [Google Scholar] [CrossRef]
- Sheka, E.F.; Golubev, Y.A.; Popova, N.A. Graphene Domain Signature of Raman Spectra of sp2 Amorphous Carbons. Nanomaterials 2020, 10, 2021. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Sung, Y.M.; Bao, N.; Tan, D.; Lee, R.; Zafra, J.L.; Lee, B.S.; Ishida, M.; Ding, J.; López Navarrete, J.T.; et al. Stable Tetrabenzo-Chichibabin’s Hydrocarbons: Tunable Ground State and Unusual Transition between Their Closed-Shell and Open-Shell Resonance Forms. Amer. Chem. Soc. 2012, 134, 14513. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Chi, C. Recent Highlights and Perspectives on Acene Based Molecules and Materials. Chem. Mater. 2014, 26, 4046–4056. [Google Scholar] [CrossRef]
- Trinquier, G.; David, G.; Malrieu, J.P. Qualitative Views on the Polyradical Character of Long Acenes. J. Phys. Chem. A 2018, 122, 6926. [Google Scholar] [CrossRef]
- Zhang, K.; Monteiro, M.; Jia, Z. Stable Organic Radical Polymers: Synthesis and Applications. Polym. Chem. 2016, 7, 5589. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Wu, Y.; Li, Y. A Review on the Origin of Synthetic Metal Radical: Singlet Open-Shell Radical Ground State? J. Phys. Chem. C 2017, 121, 8579. [Google Scholar] [CrossRef]
- Fisher, H. The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations. Chem. Rev. 2001, 101, 3581. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.G. What’s New in Stable Radical Chemistry? Org. Biomol. Chem. 2007, 5, 1321. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.G. (Ed.) Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Müllegger, S.; Rashidi, M.; Fattinger, M.; Koch, R.J. Interactions and Self-Assembly of Stable Hydrocarbon Radicals on a Metal Support. Phys. Chem. C 2012, 116, 22587. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhu, L.; Bai, R.; Lan, Y. Stabilization of Two Radicals with One Metal: A Stepwise Coupling Model for Copper-Catalyzed Radical-Radical Cross-Coupling. Sci. Rep. 2017, 7, 43579. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.X.; Li, Y.; Huang, F. Persistent and Stable Organic Radicals: Design, Synthesis, and Applications. Chem 2021, 7, 288. [Google Scholar] [CrossRef]
- Zeng, W.; Wu, J. Open-Shell Graphene Fragments. Chem 2021, 7, 358. [Google Scholar] [CrossRef]
- Radovich, L.R. Chemistry and Physics of Carbon; Radovich, L.R., Ed.; Marcel Dekker: New York, NY, USA, 2001; Volume 27, p. 131. [Google Scholar]
- Serp, P.; Figueiredo, J.L. (Eds.) Carbon Materials for Catalysis; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Serp, P.; Machado, B. (Eds.) Nanostructured Carbon Materials for Catalysis; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Radovich, L.R. Physicochemical Properties of Carbon Materials: A Brief Overview. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J.L., Eds.; Wiley: Hoboken, NJ, USA, 2009; p. 1. [Google Scholar]
- Bansal, R.C.; Donet, J.-B. Carbon Black. Science and Technology; Donet, J.-B., Bansal, R.C., Eds.; Marcel Dekker: New York, NY, USA, 1993; p. 175. [Google Scholar]
- Villa, A.; Dimitratos, N. (Eds.) Metal-Free Functionalized Carbons in Catalysis: Synthesis, Characterization and Applications; Royal Society of Chemistry: London, UK, 2018. [Google Scholar]
- Bandosz, T.J. Surface Chemistry of Carbon Materials. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J.L., Eds.; Wiley: Hoboken, NJ, USA, 2009; p. 45. [Google Scholar]
- Bel’skaya, R.I. Shungites—New Carbon Raw Materials; Karelian Science Center: Apatity, Russia, 1984. (In Russian) [Google Scholar]
- Grigoyeva, E.N.; Rozhkova, N.N. Behavior of Shungite Carbon in Reactions Simulating Thermal Transformations of Coal. Russ. J. Appl. Chem. 2000, 73, 600. (In Russian) [Google Scholar]
- Rozhkova, N.N. Perspectives of Fullerene Nanotechnology; Osawa, E., Ed.; Springer: Berlin, Germany, 2002; p. 237. [Google Scholar]
- Rozhkova, N.N.; Gorlenko, L.E.; Yemel’yanova, G.I.; Jankowska, A.; Korobov, M.V.; Lunin, V.V.; Osawa, E. Effect of Ozone on the Structure and Physicochemical Properties of Ultradisperse Diamond and Shungite Nanocarbon Elements. Pure Appl. Chem. 2009, 81, 2093. [Google Scholar] [CrossRef]
- Hu, F.; Patel, M.; Luo, F.; Flach, C.; Mendelsohn, R.; Garfunkel, E.; He, H.; Szostak, M.J. Graphene-Catalyzed Direct Friedel-Crafts Alkylation Reactions: Mechanism, Selectivity, and Synthetic Utility. Am. Chem. Soc. 2015, 137, 14473. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Dai, L. Carbon-Based Metal-Free Catalysts for Electrocatalysis beyond the ORR. Ang. Chem. Int. Ed. 2016, 55, 11736. [Google Scholar] [CrossRef] [PubMed]
- Sheka, E.F. Graphene Oxyhydride Catalysts in View of Spin Radical Chemistry. Materials 2020, 13, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapta, P.; Bartl, A.; Gromov, A.; Stasoeko, A.; Dunsch, L. Efficient and Accurate Approximations to the Molecular Spin-Orbit Coupling Operator and Their Use in Molecular G-Tensor Calculations. Chem. Phys. Chem. 2002, 3, 351. [Google Scholar] [CrossRef]
- Kosaka, M.; Ebbesen, T.W.; Hiura, H.; Tanigaki, K. Annealing Effect on Carbon Nanotubes. An ESR Study. Chem. Phys. Lett. 1995, 233, 47. [Google Scholar] [CrossRef]
- Trenikhin, M.V.; Ivashchenko, O.V.; Eliseev, V.S.; Tolochko, B.P.; Arbuzova, A.B.; Muromtsev, I.V.; Kryazhev, Y.; Drozdov, G.; Sazhina, E.; Likholobov, V.A. Electron Microscopy Investigation of Structural Transformation of Carbon Black under Influence of High-Energy Electron Beam. Fuller. Nanotub. Carbon Nanostruct. 2015, 23, 801. [Google Scholar] [CrossRef]
- Krasnovyd, S.V.; Konchits, A.A.; Shanina, B.D.; Valakh, M.Y.; Yanchuk, I.B.; Yukhymchuk, V.O.; Yefanov, A.V.; Skoryk, M.A. Local Structure and Paramagnetic Properties of the Nanostructured Carbonaceous Material Shungite. Nanoscale Res. Lett. 2015, 10, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silaev, V.I.; Ilchenko, V.O.; Lyutoev, V.P.; Filippov, V.N.; Golubev, Y.A.; Kovaleva, O.V. Problemy geologii i mineralogii (Problems in Geology and Mineralogy); Yushkin, N.P., Ed.; Geoprint: Syktyvkar, Russia, 2006; p. 283. (In Russian) [Google Scholar]
- Razumovskii, S.D.; Gorshenev, V.N.; Kovarskii, A.L.; Kuznetsov, A.M.; Shchegolikhin, A.N. Carbon Nanostructure Reactivity: Reactions of Graphite Powders with Ozone. Fuller. Nanotub. Carbon Nanostruct. 2007, 15, 53. [Google Scholar] [CrossRef]
- Cataldo, F.; Putz, M.V.; Ursini, O.; Angelini, G.; Garcia-Hernandez, D.A.; Manchado, A. A New Route to Graphene Starting From Heavily Ozonized Fullerenes: Part 3—An Electron Spin Resonance Study. Fuller. Nanotub. Carbon Nanostruct. 2016, 24, 195. [Google Scholar] [CrossRef]
- Johansson, K.O.; Head-Gordon, M.P.; Schrader, P.E.; Wilson, K.R.; Michelsen, H.A. Resonance-Stabilized Hydrocarbon-Radical Chain Reactions May Explain Soot Inception And Growth. Science 2018, 361, 997. [Google Scholar] [CrossRef] [Green Version]
- Gronert, S.; Keeffe, J.R.; More O’Ferrall, R.A. Contemporary Carbene Chemistry; Moss, R.A., Doyle, M.P., Eds.; Wiley: Hoboken, NJ, USA, 2014; p. 3. [Google Scholar]
- Piccini, G.M.; Mendels, D.; Parrinello, M.J. Metadynamics with Discriminants: A Tool for Understanding Chemistry. Chem. Theory Comput. 2018, 14, 5040. [Google Scholar] [CrossRef] [Green Version]
- Bettinger, H.F. Electronic Structure of Higher Acenes and Polyacene: The Perspective Developed by Theoretical Analyses. Pure Appl. Chem. 2010, 82, 905. [Google Scholar] [CrossRef]
- Plasser, F.; Pašalić, H.; Gerzabek, M.H.; Libisch, F.; Reiter, R.; Burgdörfer, J.; Müller, T.; Shepard, R.; Lischka, H. The Multiradical Character of One- and Two-Dimensional Graphene Nanoribbons. Ang. Chem. Int. Ed. 2013, 52, 2581. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-Y.; Narita, A.; Müllen, K. Precision Synthesis Versus Bulk-Scale Fabrication of Graphenes. Nat. Rev. Chem. 2017, 2, 0100. [Google Scholar] [CrossRef]
- Krutous, V.A. Medicinal Properties of Shungite; Shungite: Karelia, Russia, 2016. (In Russian) [Google Scholar]
- Goryunov, A.S.; Borisova, A.G.; Rozhkov, S.P. Raman Scattering of Bioconjugates of Serum Albumin and Shungite Nanocarbon. Proc. Karelian Res. Center RAS Exp. Biol. Ser. 2012, 2, 154. (In Russian) [Google Scholar]
- Kamanina, N.V.; Serov, S.V.; Shurpo, N.A.; Rozhkova, N.N. Photoinduced Changes in Refractive Index of Nanostructured Shungite-Containing Polyimide Systems. Tech. Phys. Lett. 2011, 37, 949. [Google Scholar] [CrossRef]
- Usol’tseva, N.V.; Smirnova, M.V.; Kazak, A.V.; Smirnova, A.I.; Bumbina, N.V.; Il’in, S.O.; Rozhkova, N.N. Effects of Mechanical Strength, Working Temperature and Wax Lubricant on Tribological Behavior of Polystyrene. J. Frict. Wear 2015, 36, 380. [Google Scholar] [CrossRef]
- Politi, S.; Carcione, R.; Tamburri, E.; Matassa, R.; Lavecchia, T.; Angjellari, M.; Terranova, M.L. Graphene Platelets from Shungite Rock Modulate Electropolymerization and Charge Storage Mechanisms of Soft-Template Synthetized Polypyrrole-Based Nanocomposites. Sci. Rep. 2018, 8, 17045. [Google Scholar] [CrossRef] [PubMed]
- Sadovnichii, R.V.; Mikhaylina, A.A.; Rozhkova, N.N.; Inina, I.S. The morphological and Structural Features of Quartz of Shungite Rocks of Maksovo Deposit. Trans. KarRC RAS 2016, 2, 73. (In Russian) [Google Scholar]
Figure 1.
Z→A distributions of sp2 C=C bonds in fullerene C60 (a), a fragment of (4, 4) SWCNT (b), and nanographene molecule (5, 5) NGr (c). Calculations are performed using CLUSTER-Z1 software implementing AM1 semi-empirical HF approximation [42]. Closed-shell version of the study is presented by the RHF solutions.
Figure 1.
Z→A distributions of sp2 C=C bonds in fullerene C60 (a), a fragment of (4, 4) SWCNT (b), and nanographene molecule (5, 5) NGr (c). Calculations are performed using CLUSTER-Z1 software implementing AM1 semi-empirical HF approximation [42]. Closed-shell version of the study is presented by the RHF solutions.


Figure 2.
(a). Atomic chemical susceptibility NDA of fullerene C60, distributed over the molecule atoms according to either their numeration in the output file (curve with rhombs) or in the Z→A manner (histogram). Different colors in the insert on the right distinguish six atomic groups shown by the histogram. (b) Spin density distribution.
Figure 2.
(a). Atomic chemical susceptibility NDA of fullerene C60, distributed over the molecule atoms according to either their numeration in the output file (curve with rhombs) or in the Z→A manner (histogram). Different colors in the insert on the right distinguish six atomic groups shown by the histogram. (b) Spin density distribution.

Figure 3.
Standard ACS NDA image maps over atoms of (4, 4) single-walled carbon nanotubes shown in insert with empty (curve with dots) and hydrogen-terminated (histogram) ends. The numbering follows a movement along the tube atoms from the cap to the open end.
Figure 3.
Standard ACS NDA image maps over atoms of (4, 4) single-walled carbon nanotubes shown in insert with empty (curve with dots) and hydrogen-terminated (histogram) ends. The numbering follows a movement along the tube atoms from the cap to the open end.

Figure 4.
Standard ACS NDA image maps over atoms of (5, 5) NGr molecule with empty (histogram) and hydrogen-terminated (curve with dots) edges. Insert: equilibrated structures of the parent (5, 5) NGr molecule and its monohydrogenated homologue peripentacene.
Figure 4.
Standard ACS NDA image maps over atoms of (5, 5) NGr molecule with empty (histogram) and hydrogen-terminated (curve with dots) edges. Insert: equilibrated structures of the parent (5, 5) NGr molecule and its monohydrogenated homologue peripentacene.

Figure 5.
Photodynamic effect of the [C60 + O2] complex. (a) Scheme of the IMI terms related to the interaction between two fullerene molecules. (A0B0) and (A+B−) match branches of the terms related to the interaction between neutral molecules and their ions, respectively. R+− and R00 mark minimum positions attributed to the formation of tightly bound dimer (AB) and weakly bound charge transfer complex (A0 + B0), respectively. Rscn indicates the point of avoidable intersection of terms (A0B0) and (A+B−). (b) Schematic presentation of fullerene clusterization in solution. (c) The formation of an ionic pair of C60 under photoexcitation. The only ion pair is formed under photoexcitation of the (C60)n cluster of any content. (d) Schematic presentation of the spin-flip in oxygen molecule under photoexcitation. (e) ACS distribution over atoms of (histogram) and (red curve) complexes.
Figure 5.
Photodynamic effect of the [C60 + O2] complex. (a) Scheme of the IMI terms related to the interaction between two fullerene molecules. (A0B0) and (A+B−) match branches of the terms related to the interaction between neutral molecules and their ions, respectively. R+− and R00 mark minimum positions attributed to the formation of tightly bound dimer (AB) and weakly bound charge transfer complex (A0 + B0), respectively. Rscn indicates the point of avoidable intersection of terms (A0B0) and (A+B−). (b) Schematic presentation of fullerene clusterization in solution. (c) The formation of an ionic pair of C60 under photoexcitation. The only ion pair is formed under photoexcitation of the (C60)n cluster of any content. (d) Schematic presentation of the spin-flip in oxygen molecule under photoexcitation. (e) ACS distribution over atoms of (histogram) and (red curve) complexes.

Figure 7.
Evolution of the total energy ΔH (a), molecular chemical susceptibility ND, and per-step coupling energy Ecpl (b) at growing the number of atom pairs k for C60 hydrides (dotted curves with empty circles) and C60 fluorides (solid curves with filled circles).
Figure 7.
Evolution of the total energy ΔH (a), molecular chemical susceptibility ND, and per-step coupling energy Ecpl (b) at growing the number of atom pairs k for C60 hydrides (dotted curves with empty circles) and C60 fluorides (solid curves with filled circles).

Figure 8.
Top–Equilibrium structure of the (5, 5) NGr molecule and the spatial map of the ACS NDA distribution over its atoms. (a–c) Equilibrium structures of complete graphene hydrides (left column), graphene fluoride, and two graphene oxides (right column, from top to bottom). The double notification, say, C66H44H44, means that the first 44 hydrogen atoms are bound with 22 edge atoms of the (5, 5) NGr molecule, while the other 44 hydrogens are attached to the carbon atoms located in the molecule basal plane.
Figure 8.
Top–Equilibrium structure of the (5, 5) NGr molecule and the spatial map of the ACS NDA distribution over its atoms. (a–c) Equilibrium structures of complete graphene hydrides (left column), graphene fluoride, and two graphene oxides (right column, from top to bottom). The double notification, say, C66H44H44, means that the first 44 hydrogen atoms are bound with 22 edge atoms of the (5, 5) NGr molecule, while the other 44 hydrogens are attached to the carbon atoms located in the molecule basal plane.

Figure 9.
Appearance of natural amorphous carbon (top) and equilibrium structures of molecular BSU models (middle): (a) graphene oxihydrides C68O4H6 attributed to “C=O” shungite carbon, (b) C64O4H10 of “O=C-O-C’ antraxolite, (c) and (d) C64O4H3 (c) and C64O4 (d) of “C-O-C” carbon blacks CB632 and CB624, respectively. (bottom) NDA distribution over atoms of BSU model molecules (see text). UHF calculations.
Figure 9.
Appearance of natural amorphous carbon (top) and equilibrium structures of molecular BSU models (middle): (a) graphene oxihydrides C68O4H6 attributed to “C=O” shungite carbon, (b) C64O4H10 of “O=C-O-C’ antraxolite, (c) and (d) C64O4H3 (c) and C64O4 (d) of “C-O-C” carbon blacks CB632 and CB624, respectively. (bottom) NDA distribution over atoms of BSU model molecules (see text). UHF calculations.

Figure 10.
Equilibrium structures of (SiO4)48 (a) and C66H6O4 graphene molecule (b) imitating α-quartz nucleus and BSU of shungite carbon, respectively. (e,f) Equilibrium structures of silica–shungite carbon composites at starting positions of the components (c) and (d), respectively. UHF calculations.
Figure 10.
Equilibrium structures of (SiO4)48 (a) and C66H6O4 graphene molecule (b) imitating α-quartz nucleus and BSU of shungite carbon, respectively. (e,f) Equilibrium structures of silica–shungite carbon composites at starting positions of the components (c) and (d), respectively. UHF calculations.


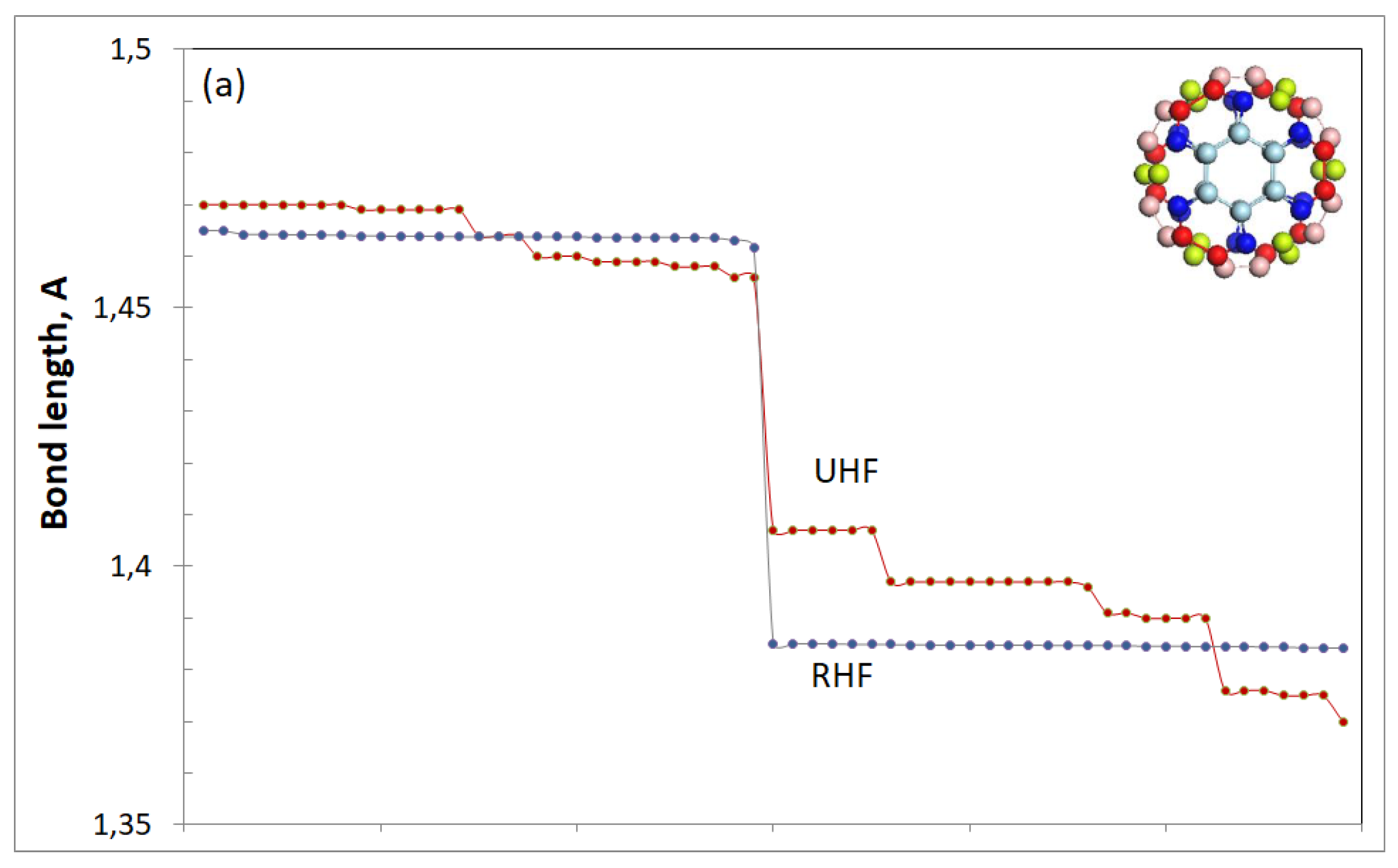{kind=link}
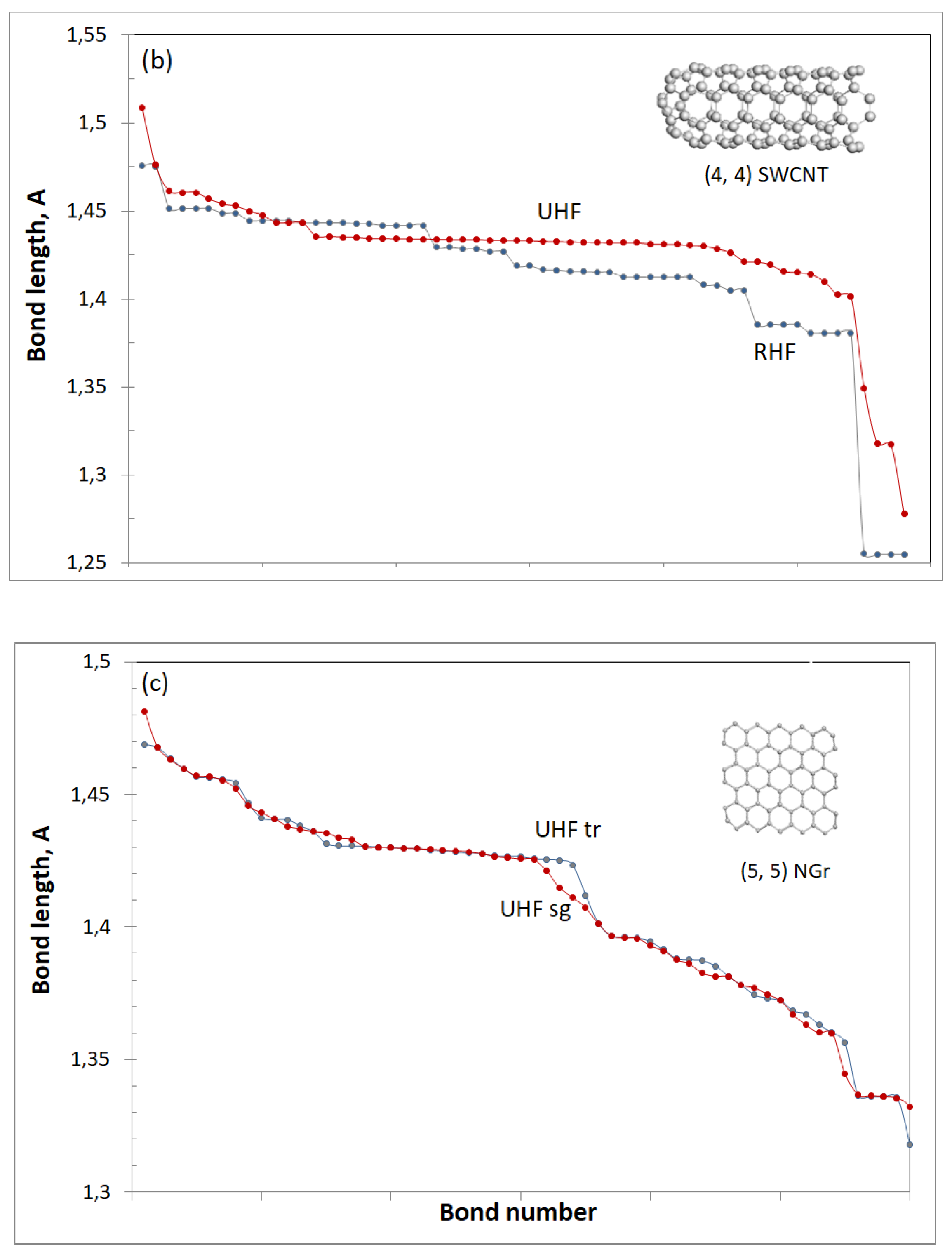{kind=link}
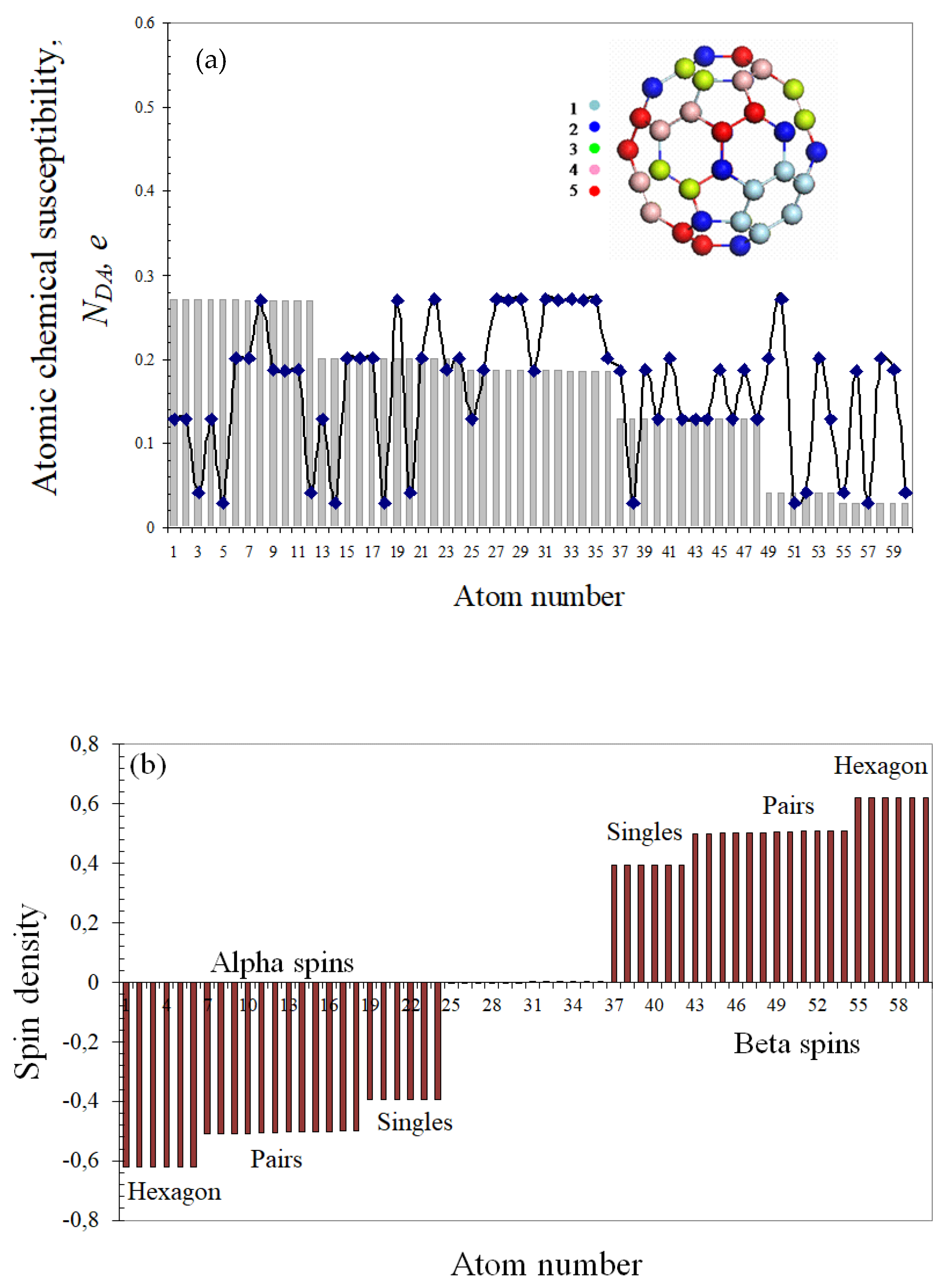{kind=link}
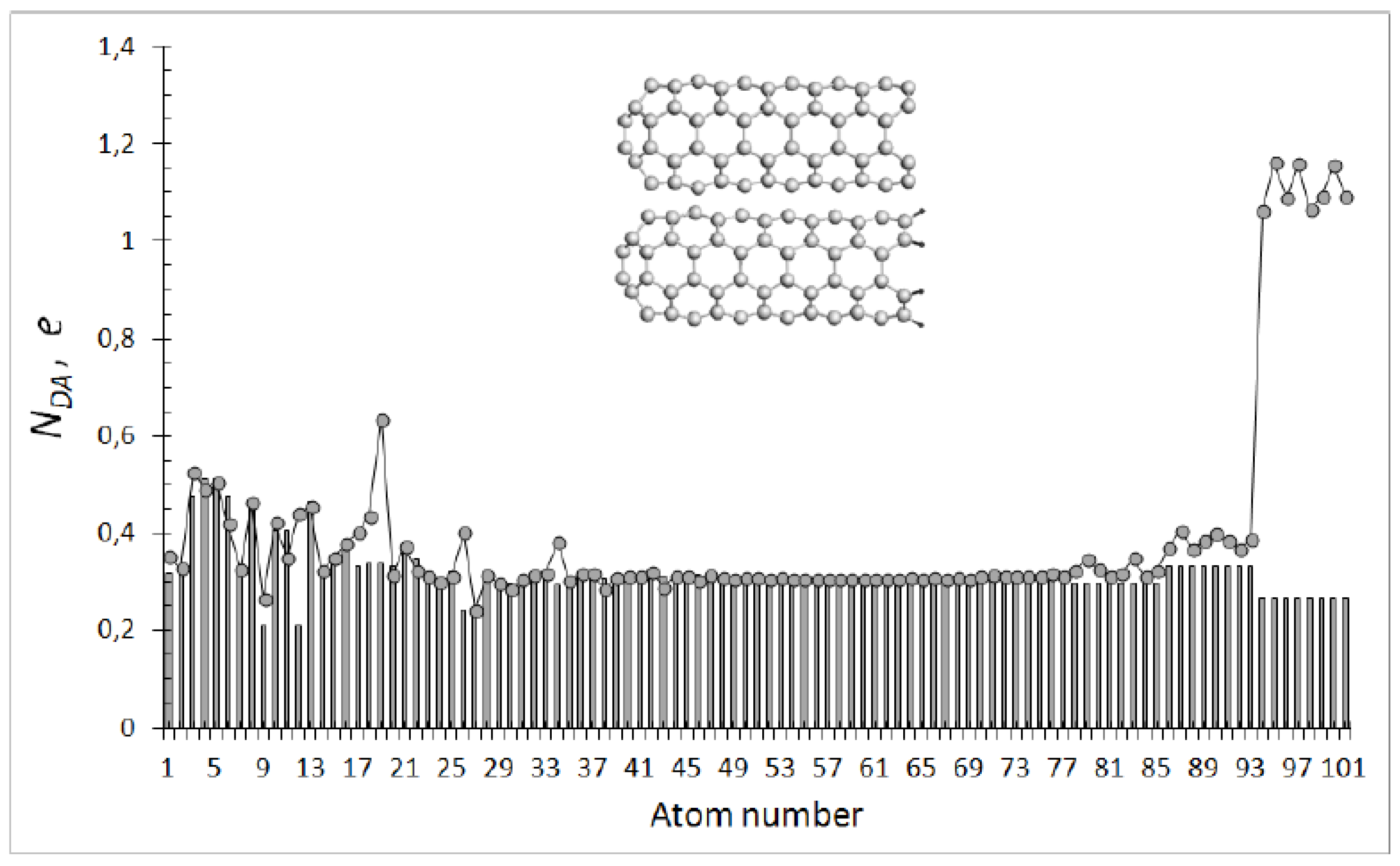{kind=link}
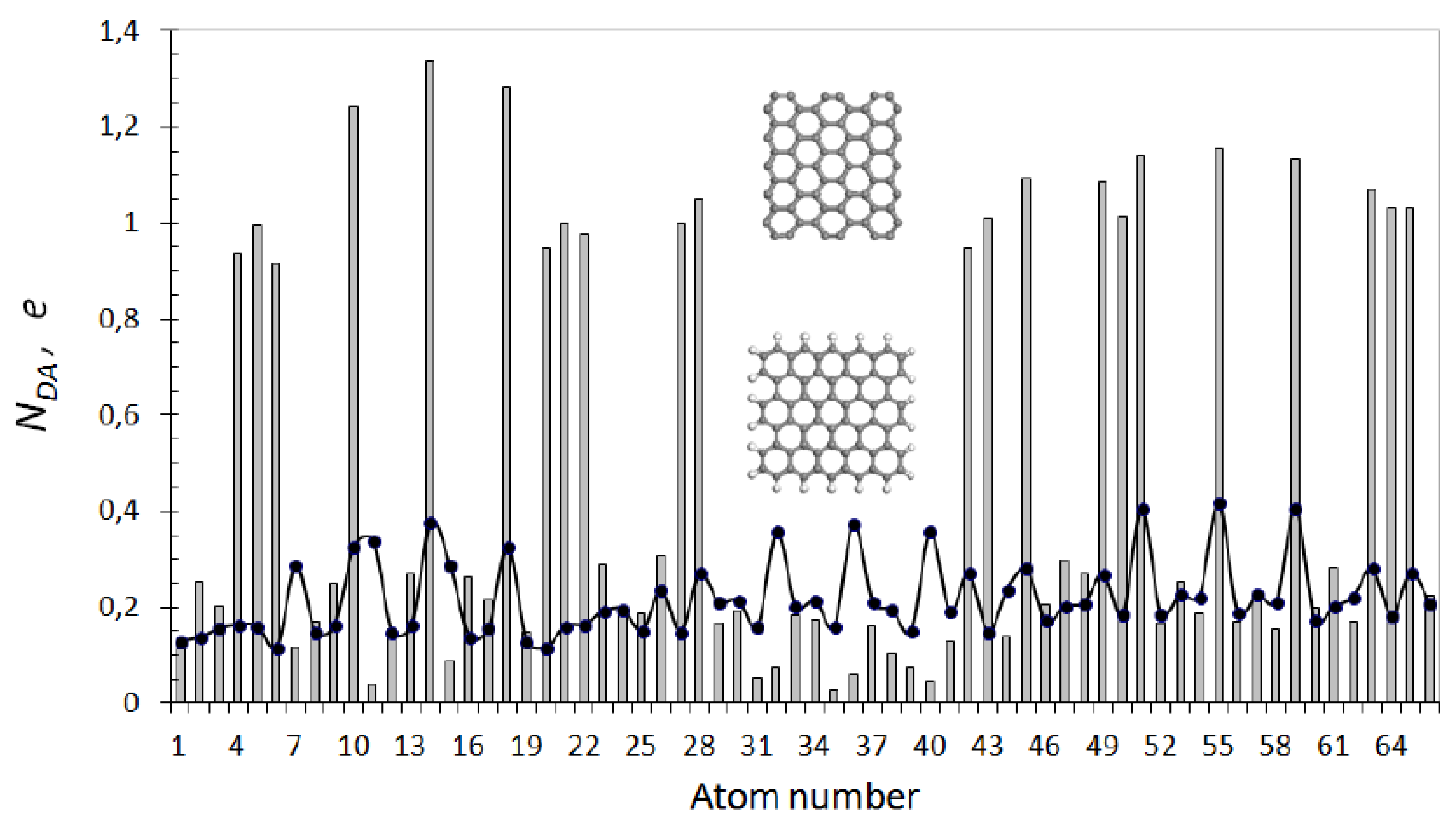{kind=link}
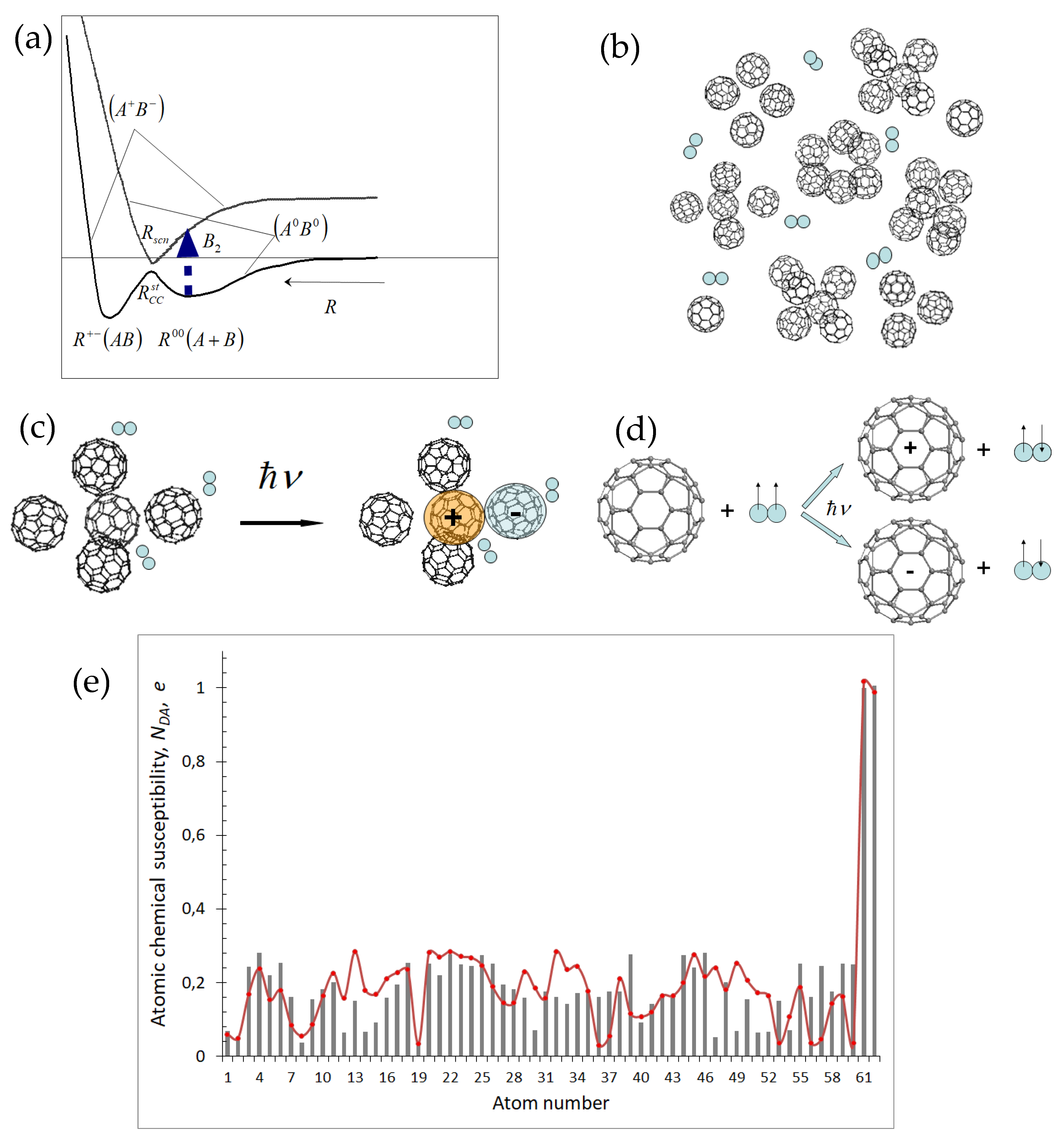{kind=link}
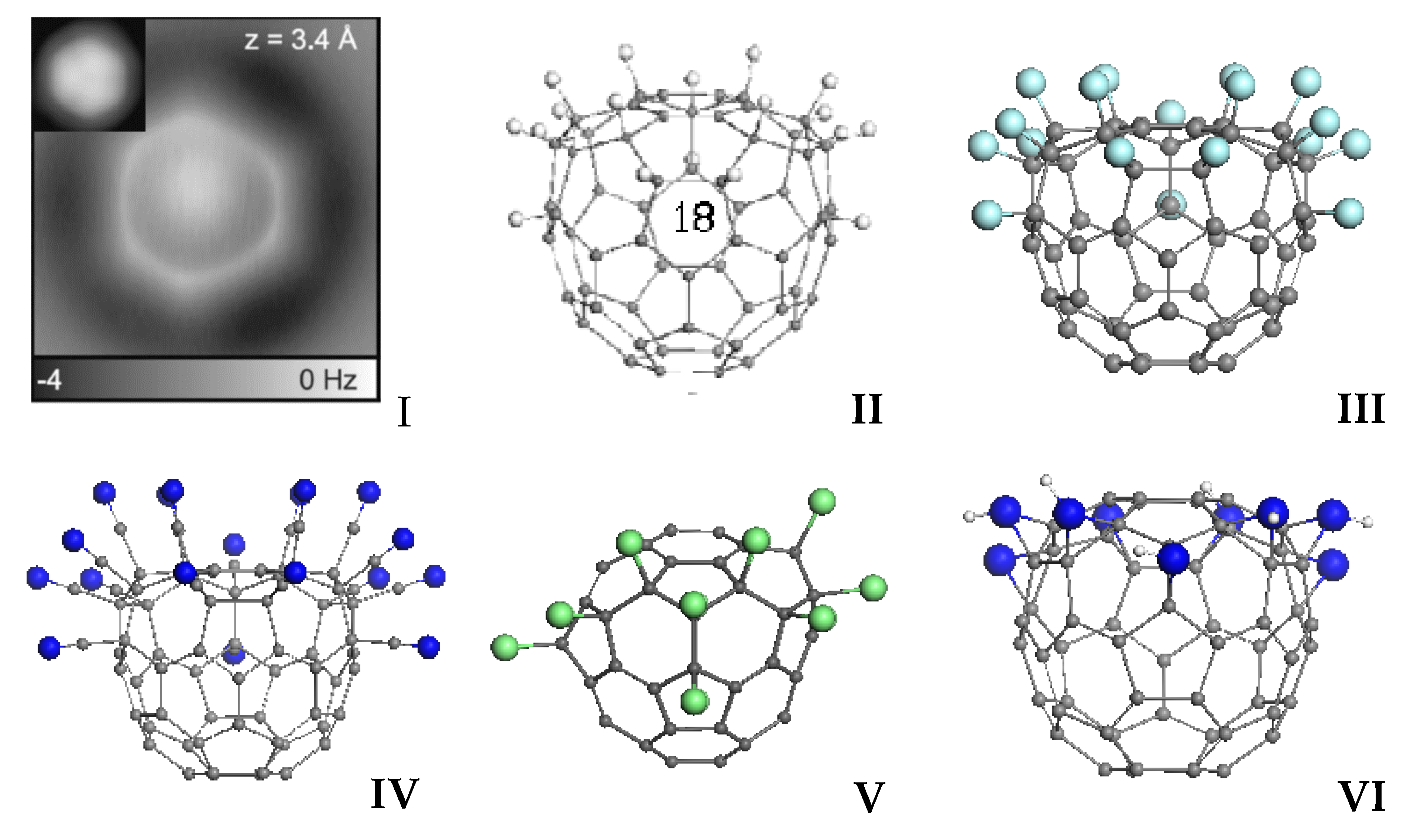{kind=link}
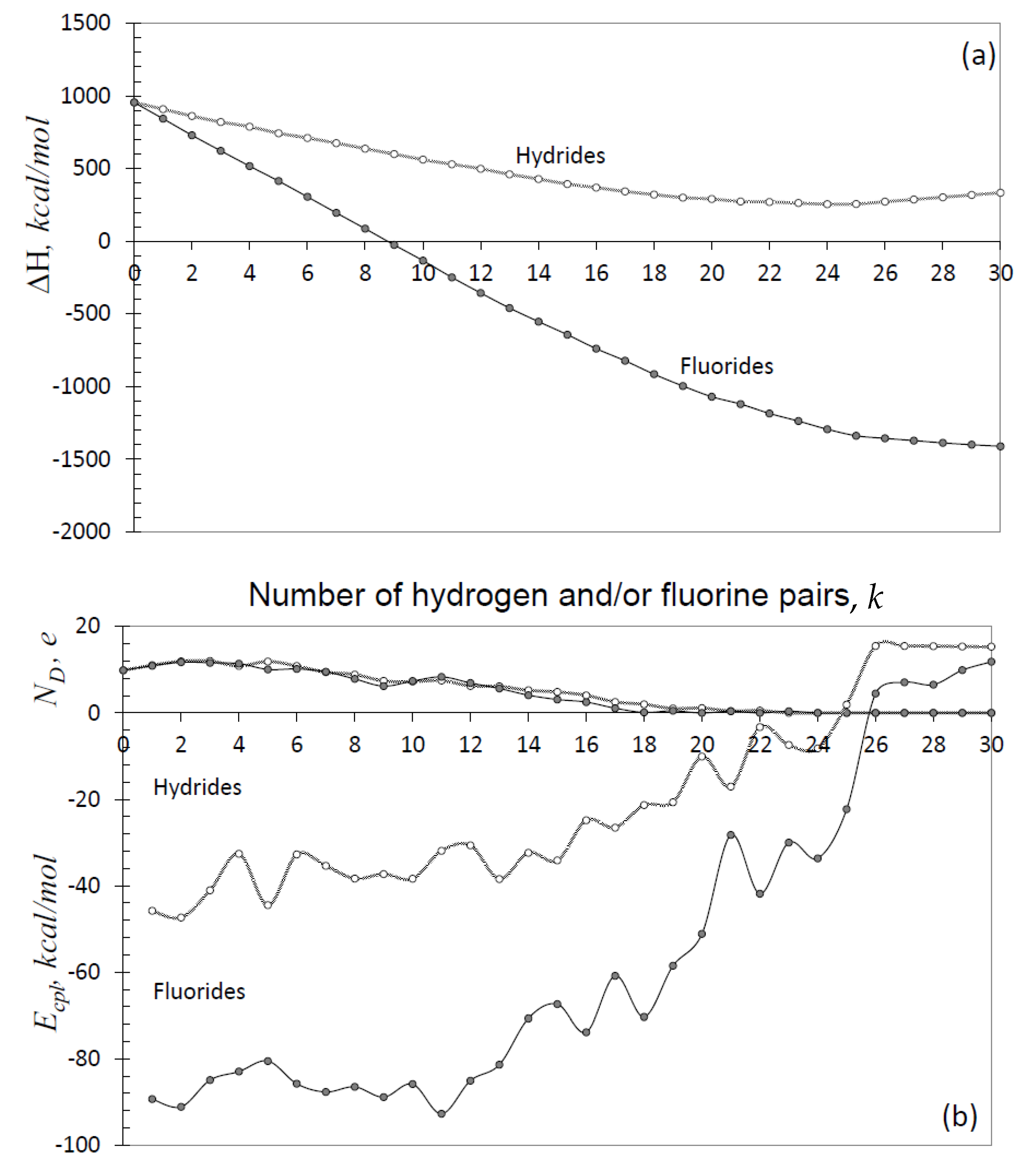{kind=link}
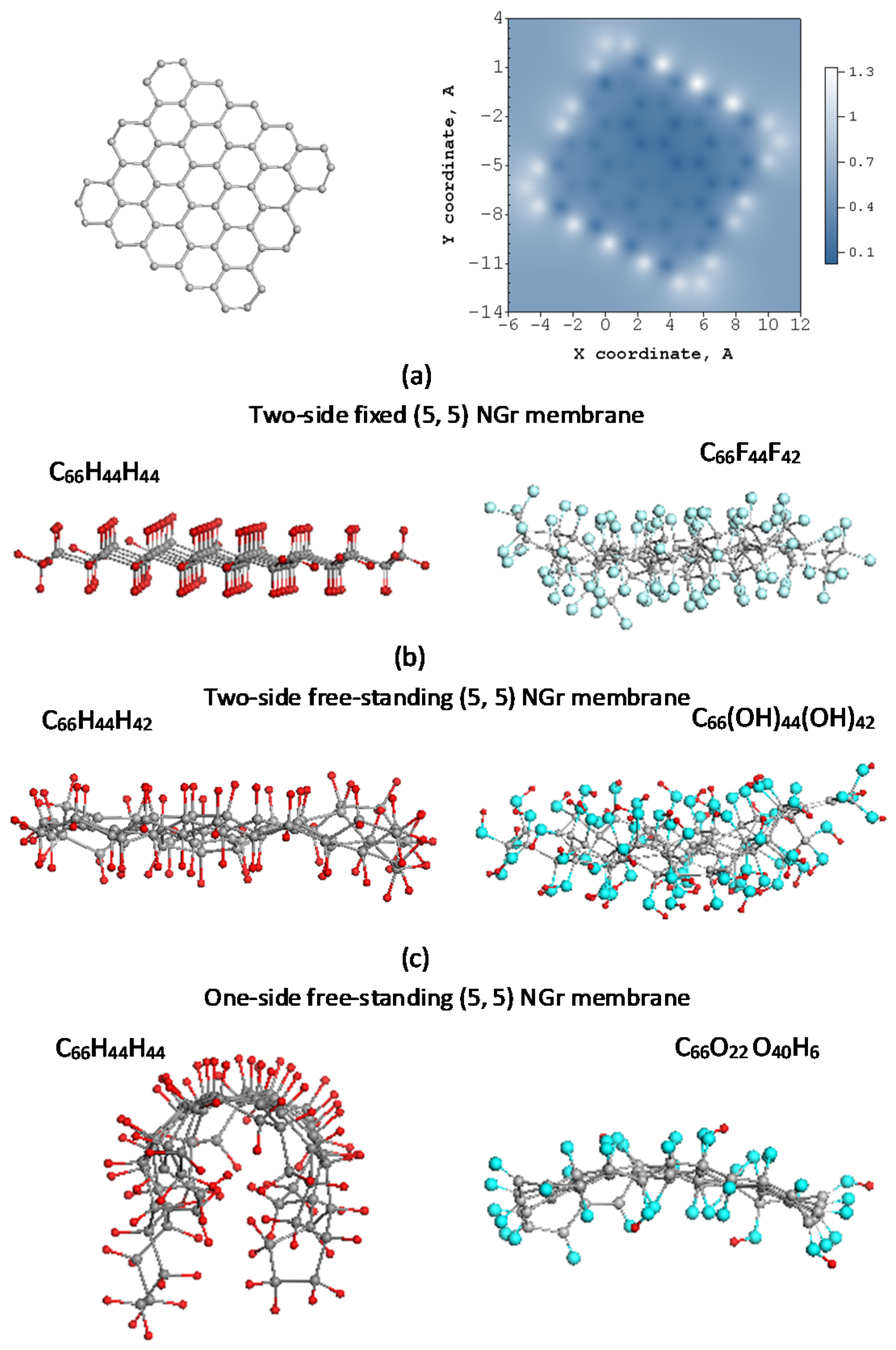{kind=link}
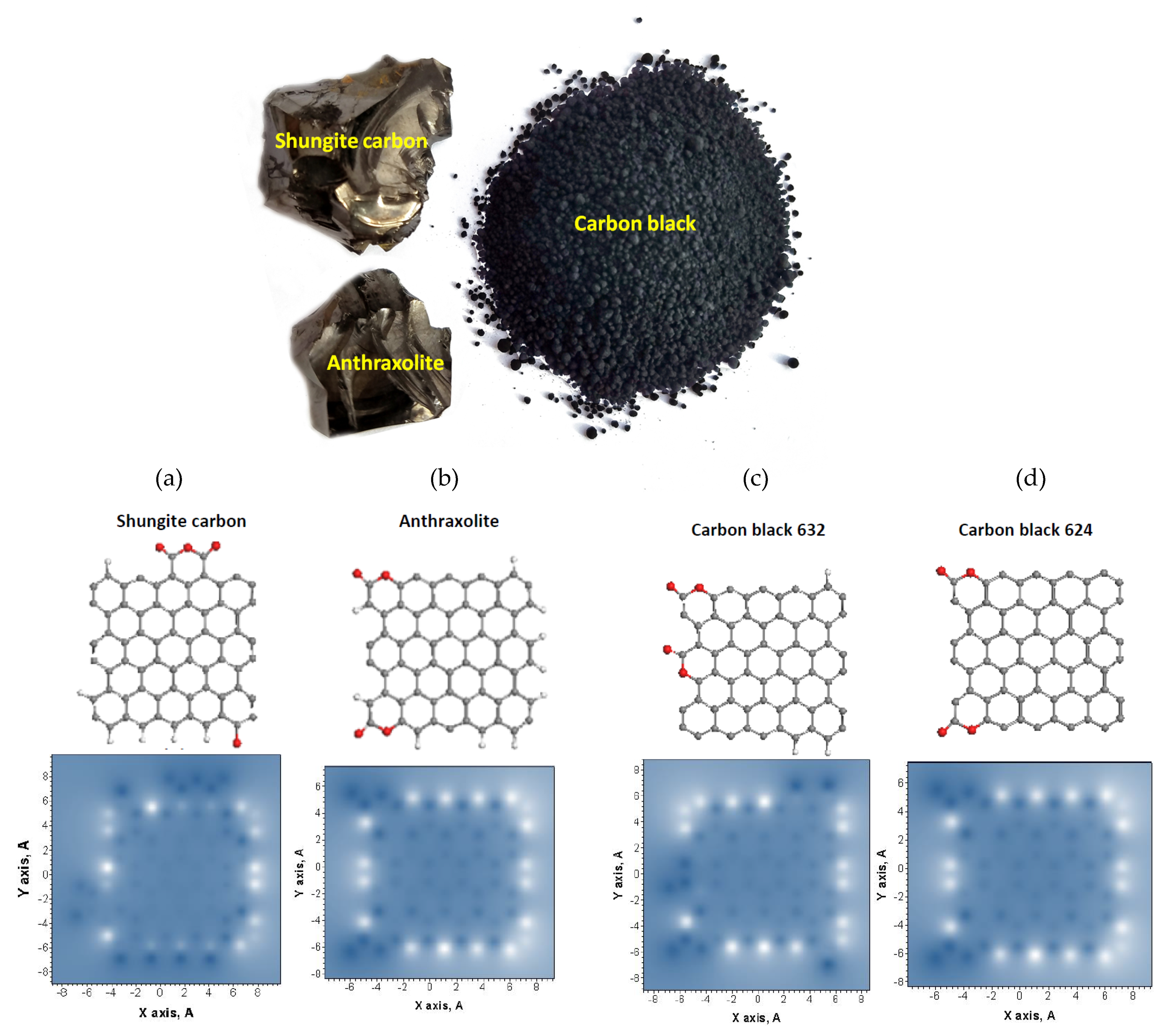{kind=link}
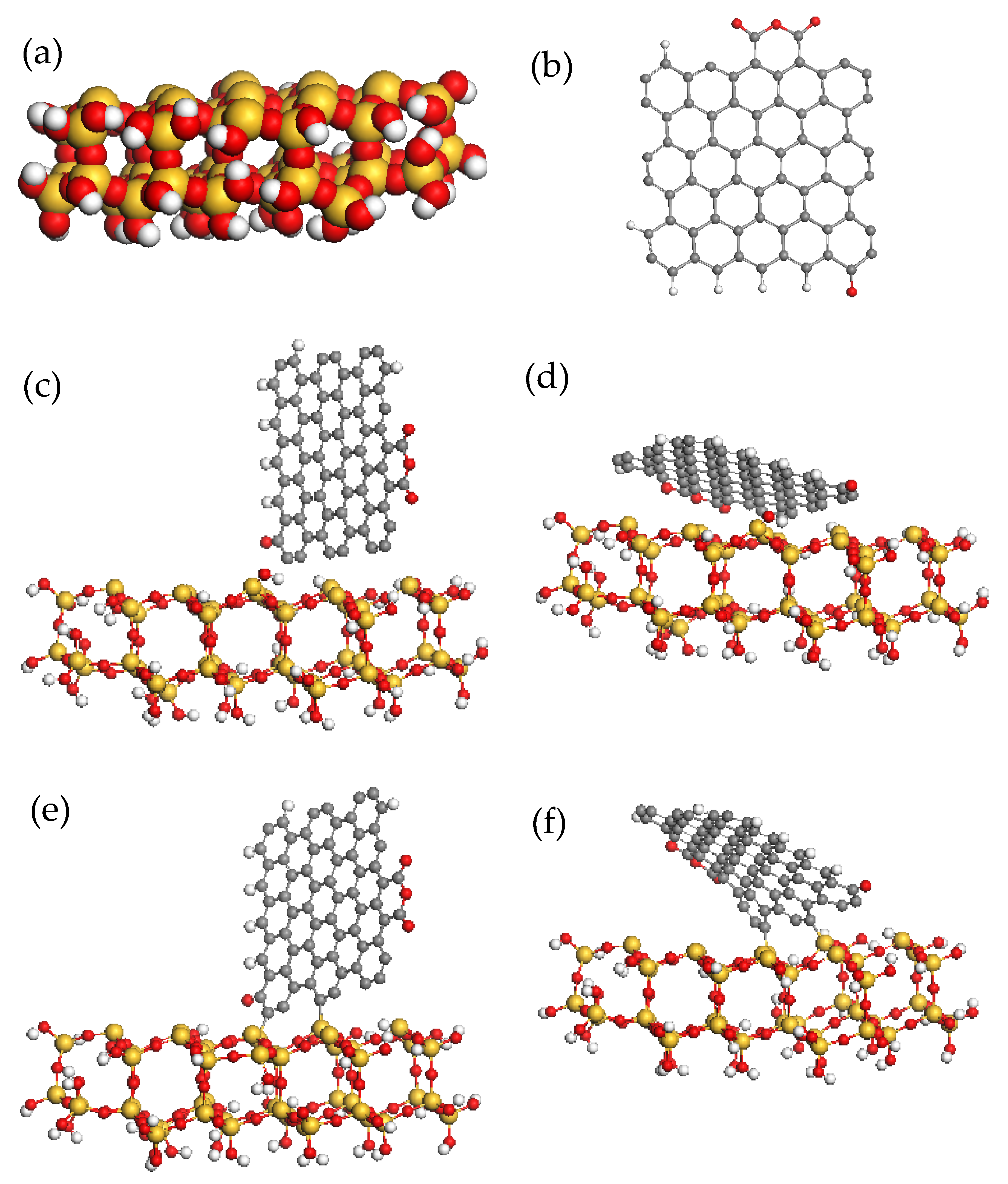{kind=link}
Table 1.
Energies (kcal/mol) and emergent spin characteristics of the sp2 molecules in the ground state.
Table 1.
Energies (kcal/mol) and emergent spin characteristics of the sp2 molecules in the ground state.
| Molecules | ||||||||
|---|---|---|---|---|---|---|---|---|
| C60 | 970.180 | 955.362 | 14.818 | 982.917 | 27.555 | 4.92 | 0 | 9.84 |
| (4, 4) SWCNT | 1819.509 | 1648.338 | 171.171 | 1627.308 | −21.030 | 19.758 sg | 0 | 39.515 sg |
| 20.325 tr | 2 | 38.651 tr | ||||||
| (5, 5) NGr | 1802.381 | 1454.492 | 347.889 | 1448.396 | −6.096 | 16.930 sg | 0 | 33.860 sg |
| 18.121 tr | 2 | 34.242 tr |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sheka, E.F. sp2 Carbon Stable Radicals. C 2021, 7, 31. https://doi.org/10.3390/c7020031
AMA Style
Sheka EF. sp2 Carbon Stable Radicals. C. 2021; 7(2):31. https://doi.org/10.3390/c7020031
Chicago/Turabian StyleSheka, Elena F. 2021. "sp2 Carbon Stable Radicals" C 7, no. 2: 31. https://doi.org/10.3390/c7020031
APA StyleSheka, E. F. (2021). sp2 Carbon Stable Radicals. C, 7(2), 31. https://doi.org/10.3390/c7020031
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.