1. Introduction
Presently, petroleum-derived cokes (Asbury Carbons, PetCoke—
https://asbury.com/materials/coke/ (accessed on 3 December 2021)), coal tar pitches and mined (i.e., “natural”) graphite are the primary precursors for graphitic carbons [
4,
5], while oil-based synthetic resins and polymers are preferred precursors for manufactured carbon materials for their graphitic quality and uniformity [
6,
7,
8]. Unfortunately, petroleum-derived cokes and coal tar pitches are nonrenewable resources that require intensive energy for their production (American Energy Reliance (AER)—Introduction to Petroleum Coke
https://www.americanenergyalliance.org/2014/02/introduction-to-petroleum-coke/ (accessed on 3 December 2021); Repsol, Petcoke production
https://www.repsol.com/en/products-and-services/green-fuel-grade petcoke/production/index.cshtml (accessed on 4 December 2021)). Moreover, they are accompanied by high organic volatile emissions. Meanwhile, natural graphite reserves are limited and the mining process is environmentally poor (Whoriskey, P., In your phone, in their air. A trace of graphite is in consumer tech. In these Chinese villages, it’s everywhere. 2 October 2016.
https://www.washingtonpost.com/graphics/business/batteries/graphite-mining-pollution-in-china/ (accessed on 3 December 2021); The unseen side of graphite mining. 6 December 2011.
http://rukshanmaliq.blogspot.com/2011/12/unseen-side-of-graphite-mining.html (accessed on 3 December 2021)).
Bio-based, renewable precursors such as cellulose would displace the oil and mined graphite as presently used for the manufacture of carbons for energy storage or composites [
9,
10,
11]. Water usage, as used in oil production and processing, and CO
2 emissions would be reduced. Yet, when lignocellulosic biomass and other carbohydrate-based or oxygen-rich precursors are carbonized, the formation of non-hexagonal carbon ring structures (pentagons, heptagons, and octagons) within graphene-like sheets prevents graphitization [
12,
13]. The resulting disordered carbon lacks the galleries formed between parallel and stacked lamellae. Moreover, the electrical conductivity of these carbons is also poor when compared to graphitic carbons [
14,
15,
16]. Templating offers a path to convert a non-graphitizing bio-based precursor, such as sugar, into a graphitizing carbon.
Relevant to interaction with the matrix, distinctive chemical features of graphene-oxide (GO) and reduced graphene-oxide (rGO) are the sp
2 framework and oxygen content. Physical templating is defined as the interaction between the emerging aromatic domains within the matrix and the sp
2 framework of the GO leading to alignment and orientation. Reactive templating is defined as chemical bond formation between the matrix and GO. Bonding would be favored by either or both components being in radical form, namely, matrix decomposition products or radical sites on GO (or rGO) generated by loss of CO or CO
2. The different oxygen contents for GO versus rGO affect both its physical and chemical templating ability. High oxygen content will lead to extensive radical sites at elevated temperatures. Oppositely, high oxygen content disrupts the sp
2 framework with the conversion of many sp
2 basal sites into sp
3 carbons supporting phenolic and epoxide groups [
17,
18] and the size of contiguous sp
2 domains is reduced.
These definitions are supported by reactive force field (ReaxFF)-based molecular dynamics (MD) simulations. These simulations reveal that the addition of graphene to polyacrylonitrile (PAN) precursor enhances the alignments of 6-member carbon rings in carbonization [
19,
20], with π-π interactions presumably being the origin of the alignment force. Related simulations for double-walled nanotubes and graphene within graphitizing PAN showed that the alignment between the template surface and newly formed all-carbon rings improves continuously during the carbonization process by providing a stable sp
2 basal surface [
21]. MD simulation trajectories have also revealed that graphene surface carbon atoms are relatively inactive and do not form bonds with the polymer matrix, even at high temperatures, due to their conjugated sp
2 electronic configuration [
20,
22]. Instead, C-H bonds at the graphene edges break and the undercoordinated carbon atoms at the graphene edges then form bonds with PAN chains. Here, we test oxygen groups as the labile entity to create reactive edge sites.
These concepts are supported by experimental studies. For example, addition of a small amount of GO into PAN increased graphitic order and preferred orientation in the vicinity of visible GO [
23]. A separate study found that, during the thermal stabilization of PAN/rGO, rGO confined the cyclization and dehydrogenation reaction of PAN to facilitate the formation of large, conjugated structures [
24]. In a related study, graphene was shown to induce carbonization of polyimide [
25]. Films doped with 2 wt.% GO were found to have higher crystallization with lamellae orientation and interlayer distance closer to that of ideal graphene compared to that doped with 2 wt.% rGO. This was attributed to bonding between the GO hydroxyl and carboxylic groups and PAN. Elsewhere, PAN/GO composite films spin-coated on substrates were stabilized in air and then carbonized under inert atmosphere at 1000 ℃ to form carbon films [
26]. Cyclization of PAN and partial reduction of GO occurred simultaneously during stabilization. High electrical conductivity appeared due to the π-π interaction between the graphene planes and the cyclized PAN structures [
27]. Using SU-8, a photoresist polymer, as host matrix, at the early stages of carbonization, graphene induced the carbon atoms in its near vicinity to form oriented layers parallel to the graphene layer [
28]. Upon graphitization, graphene narrowed the distribution of graphite grain orientations.
Here, in contrast to these graphitizable matrix precursors, sugar was chosen as the precursor for its well-known non-graphitizable char as an exacting test of templating to form graphitic carbon—our first objective. As a uniform mixture, the potential for crystallite formation and growth could be probed beyond the additive-matrix interface—the second objective. Last, as a simple disaccharide, results with sugar serve as a reference for more complex, but chemically equivalent, biopolymers, such as starch and cellulose—the third objective of this study.
2. Materials and Methods
2.1. Materials
GO and rGO were used as received from Cheap Tubes Inc. (Grafton, VT, US). GO has 45~55 atomic % oxygen while rGO exhibits ~5 atomic % oxygen, as specified by the manufacturer from XPS measurements. X-Y for both materials is in the 300–800 nm range, each with 2–4 layers stacked for a thickness of ~1.2 nm. rGO is produced from the same graphene oxide, hence, the nominal sheet sizes and stacking are equivalent. XPS analysis by the vendor did not report metal contamination, and our own results did not reveal any Mn or Fe contamination to the 0.1 at.% level. TGA-MS studies have shown similar profiles, i.e., CO and CO
2 gasification products form at similar temperature ranges and in amounts traceable to functional group identity [
29,
30]. From ambient to 550 ℃, there is a progressive loss of oxygen in the form of CO and CO
2. Based on these studies ~50–70% of the oxygen has been transformed into CO and CO
2 for both graphene oxide forms. Each form (rGO and GO) will lose roughly the same fraction of oxygen, albeit different absolute amounts. Food grade sugar (chemically, sucrose) was purchased from Sigma-Aldrich (St. Louis, MO, US).
2.2. Formulation
To synthesize the composites, 2.5 wt.% of a graphene form is thoroughly mixed with the matrix material, both in powdered form. These are mixed to a visible uniform dispersion of the filler in the matrix. The mixture is transferred to an aluminum mold, which is placed inside a closed reactor under autogenic pressure conditions of ¾” internal diameter and 25 mL internal volume. The mold fits into the inner dimensions of the tubing reactor. The reactor is then sealed and purged of any oxygen with an ultra-high purity nitrogen manifold. Pressurized nitrogen helps check for air leaks from the reactor tube and helps purge it of any air/oxygen. The reactor is purged 4–5 times; a slight nitrogen overpressure of ~5 psi is maintained in the reactor to ensure an inert atmosphere during the heating process.
Carbonization: Once sealed and purged, the tubing reactors were plunged into a pre-heated and aerated sand bath. The temperature of the sand bath was set at 550 °C and was maintained isothermally for a duration of ~ 5 h. This temperature (550 °C) may vary, based on the rates of carbonization of the matrix material used. Most matrices used in this work carbonize in the range of 550–650 °C for 5–6 h. The reactors oscillated to provide vertical agitation over a 2.5 cm amplitude and at a frequency of 100 oscillations per minute for the entire duration of the heat treatment process. Carbonization occurs under autogenous pressure; pressures of up to 1500–2000 psi were reached at the end of the process. At the end of carbonization, the reactors were quenched in cold water for ~5 s. Once cool, the intermediate carbonized form of the composite was retrieved after venting out released gases in a fume hood. The material was washed with acetone and left to dry overnight in a fume hood before graphitization.
Graphitization: This is the next step in the formulation process. Here, the carbonized product was held in graphite crucibles and heated to ~2700 °C in Argon (under a 2 psi overpressure) in a Centorr Vacuum Industries (Nashua, NH, US) series 45 graphitization furnace. The furnace was purged of any oxygen by flushing the furnace chamber with Argon down to 100 millitorrs at least three times before heating. Temperatures up to 1400 °C were measured with a K-type thermocouple, and a Mirage Ircon pyrometer was used to measure higher temperatures up to 2700 °C using a viewport. Samples were held at 2700 °C for ~1 h, after which the furnace and samples were cooled overnight before retrieving them for further characterization.
2.3. Material Characterization
Transmission electron microscopy: Transmission electron microscopy (TEM) and its high-resolution version (HRTEM) was performed using a FEI (Hillsboro, OR, US) Talos F200X equipped with a 200 keV FEG source with a resolution of 0.12 Å. C-C composite samples were ground and dispersed in methanol. The sample was sonicated using a sonication horn for about ~5–10 min before being dropped onto 300 mesh C/Cu lacey TEM grids. Samples were also characterized by selected area electron diffraction (SAED) patterns performed with the same electron microscope.
X-ray Diffraction: X-ray diffraction (XRD) measurements of the samples were carried out in a PANalytical (Malvern, UK) Empyrean X-ray diffractometer using a Cu source (λ = 1.54 Ǻ), para-focusing optics, and a PIXcel 3D detector. Spectrum was collected from 2θ = 5–95°. Measurements were performed after correcting for instrument broadening using an internal silicon standard. Background subtraction and peak deconvolution of the XRD pattern was performed using JADE
® (Christchurch, New Zealand) software, which also calculated the d
002 layered plane spacing as per the Bragg equation. For the sugar (matrix only) and rGO + sugar composite, the two d
002 values correspond to peaks obtained by deconvolution. The carbon layer plane lateral extent (La) and stack height (Lc) were calculated by applying the Scherrer equation [
31].
Several dozen TEM images were acquired across the samples. Multiple samples were prepared and 2–3 widely separated areas on each grid were examined. Given that the material was formed and processed as a powder, there was no control of particle (and crystallite) orientation. Images were selected as representative but not intended to be definitive for graphitic structure formation. XRD was performed on a ZBI silicon plate holding 50 mg of powder. With powder at ~+50 mesh, the material was deemed sufficiently “fine” to uniformly present crystallites in all orientations for unbiased spectra by our prior experience with such powder forms of graphitized carbons. The XRD instrument, an Empyrean III, did not allow for sample rotation
TEM and XRD are complementary techniques, where TEM/HRTEM is useful for illustrating the development of extended, stacked lamellae, while XRD offers a “bulk” measurement over a larger sample. One must exercise caution while discerning lattice spacing from bright field TEM images. This approach is risky given focusing adjustments at any given magnification. SAED patterns are preferred for lattice measurements, as they are not subject to focusing. As shown further, XRD and HRTEM measurements and characterization were consistent and complimentary.
3. Results
Figure 1 shows an HRTEM image of the matrix, sugar, along with its corresponding strong ring-like diffraction pattern, when it was subjected to high-temperature heat treatment by itself, without any added fillers. Sugar is a non-graphitizing (Here, in the context of the TEM images, “non-graphitizing” refers to ribbons, curved lamellae and voids observed, along with absence of ordered stacking.) carbon and its nanostructure exhibits this with multiple intertwined few layer ribbons visible corresponding to the strong ring-like SAED pattern. It indicates the presence of many randomly oriented crystals in the material, with no long-range order, characteristic of a non-graphitizing material.
The following panel in
Figure 2 shows GO and rGO when heat treated by themselves, as well as when their respective composite materials were formed with GO and rGO as fillers, respectively, in sugar as the surrounding matrix. GO and rGO possess similar starting sheet-like nanostructure, with rGO derived from GO, its oxidized counterpart. On being added as fillers within the sugar matrix and subjected to high-temperature heat treatment, both materials showed very different nanostructures. GO shows a sheet-like nanostructure, while rGO contained a mix of inter-locked ribbons and graphitic sheets, as discussed and shown further.
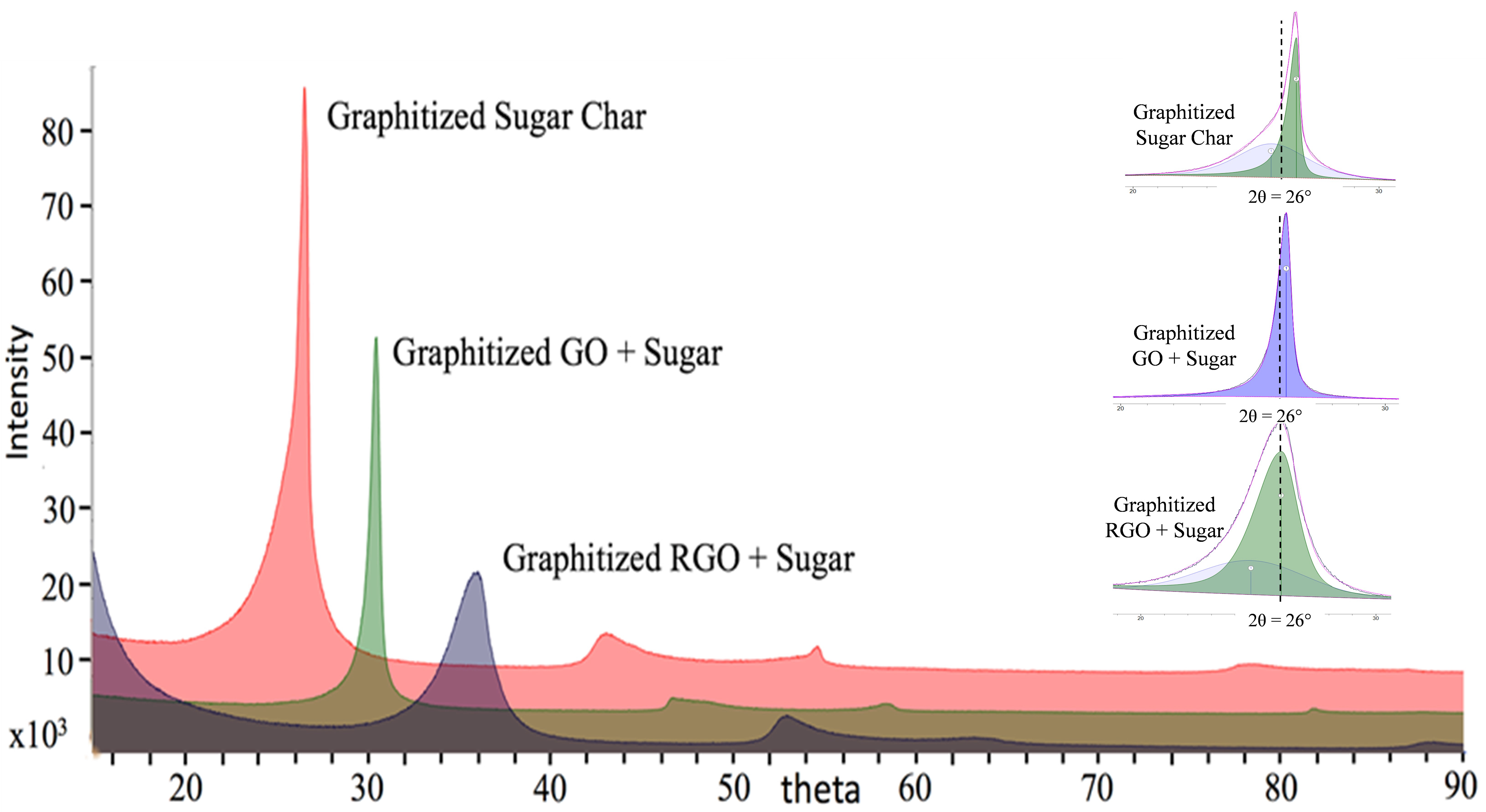
Figure 3 shows comparative XRD spectra for sugar, a well-known non-graphitizing carbon, and two composites, one with rGO + sugar, the other with GO + sugar, each for the same added 2.5 wt.%. The key to this comparison is that the rGO was produced from the same graphene oxide; hence, the nominal sheet sizes and stacking are equivalent. The relatively smaller diffraction angle of the pure sugar is characteristic for the curved lamellae, possessing larger separation, as illustrated by the HRTEM image below. The XRD breadth reflects the low stacking order in the material.
Lattice and crystallite parameters for the sugar and GO composites are summarized in
Table 1. Overall results are quantified by the graphitization index, a metric based on the lattice d-spacing, as extracted from XRD spectra. The limits are 1 for a graphitic material and 0 for a non-graphitic material. (There can be a slight negative value for certain materials. This is due to the limits of the definition). The result is telling—the lower amount of graphitic additive (GO) produces a more graphitic composite.
As shown in the inset of
Figure 3 and
Figure 4, deconvolution of the main d
002 peak for the rGO + sugar composite reveals two d
002 peaks, one broader, appearing at a lower (mean) angle of 24° and a much sharper peak, appearing at 26°, representing a smaller d
002 spacing and corresponding to higher stacking order than the former by Scherrer’s analysis. The broader peak is consistent with a non-graphitized, sugar-derived carbon, while the latter is indicative of a very graphitic phase in the composite.
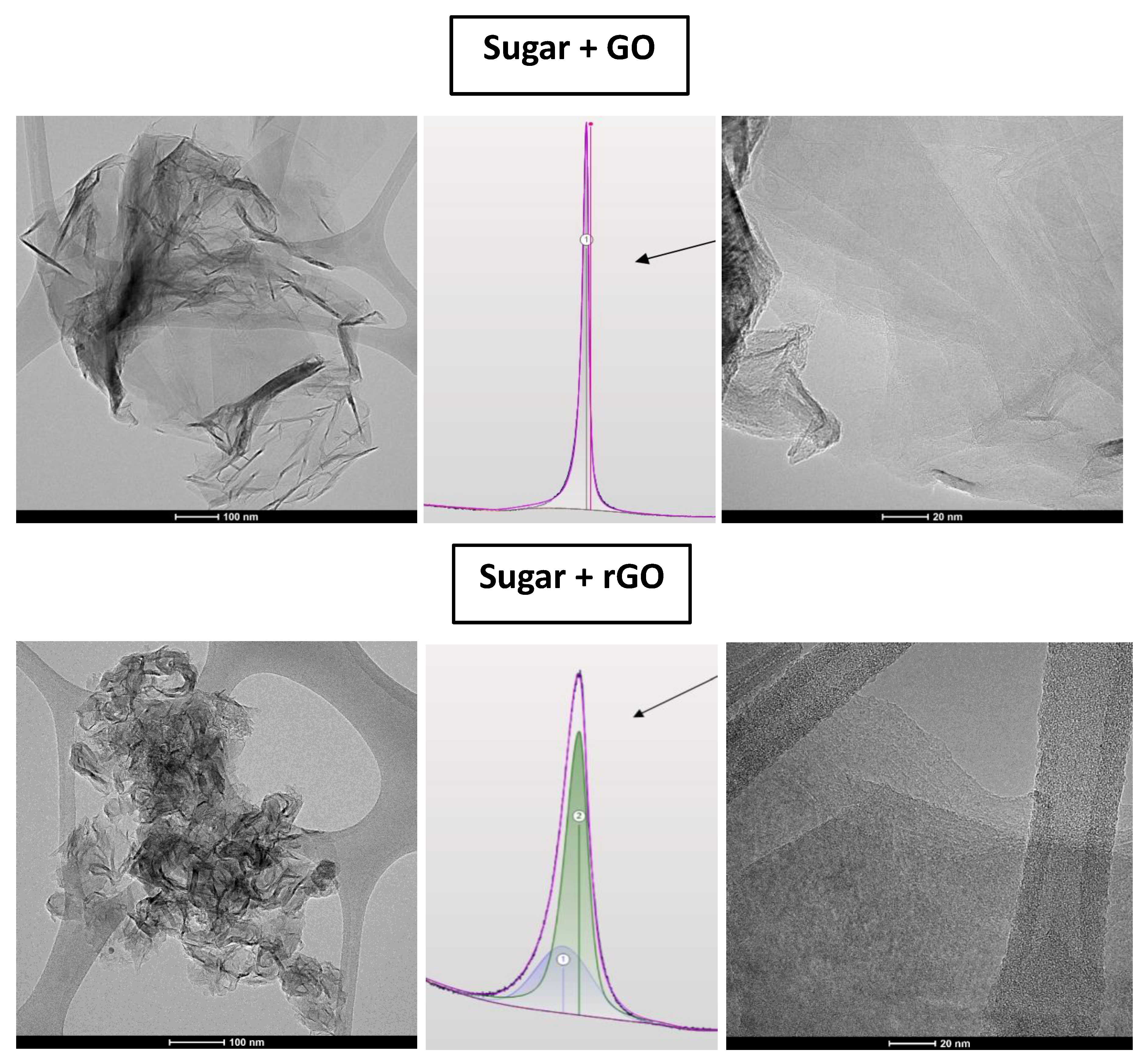
The existence of the two phases is also supported by HRTEM images and their corresponding SAED patterns shown in
Figure 5. A clear spot pattern is indicative of long-range order and crystallinity in one direction, while an intermediate spot-ring pattern indicates many randomly oriented crystal structures, resulting in their respective spot patterns merging into a ring-like pattern. This is consistent with reactive templating, as discussed further. GO + sugar, in contrast, showed only one uniform phase throughout, as shown in
Figure 6, as well as in prior HRTEM images of the composite in this manuscript.
4. Discussion
The graphitic phase component for the rGO + sugar is some fraction of the composite that is highly graphitic whereas the rest is not, being normal, sugar-derived char as is historically known. In contrast, the sugar char with GO exhibited a single (002) diffraction peak, signaling a single graphitic phase. Comparing rGO and its precursor, GO, in a non-graphitizing matrix, demonstrates reactive templating. The reduced form with largely restored basal plane carbons contained few remaining oxygen groups to form free radical sites during carbonization. As a result, rGO is largely unable to reactively bond with the matrix and exert reactive templating. Given the weak interaction between an extended aromatic π network and a non-aromatic (sp3) precursor (sugar), the possibility for physical templating appears to be minimal.
In contrast, GO forms many radical sites that will seek termination by reactive bonding with radical sites within the decomposing matrix. These comparative results demonstrate reactive bonding between “like” additive and matrix with similar chemistries and oppositely, little templating when allotrope additive and matrix are dissimilar i.e., mismatched in their chemistry. By this comparison, the limiting “factor” for rGO as an additive to induce graphitization (through reactive templating) is its residual oxygen content after reduction from the GO form.
Therein, the graphitic component for the rGO composite and the GO composite both arise through, and are illustrations of, reactive templating, wherein radical sites (mainly on the sheet edges) formed by oxygen group loss during carbonization bond with the numerous free radicals formed from the sugar matrix during thermal decomposition.
Figure 5 and
Figure 6 show HRTEM images for the rGO and GO composites, in which the graphitic and uniform phase for the GO composite is apparent, whereas rGO, by comparison, reveals both graphitized and non-graphitized carbon regions. Notably, the two phases visible for the rGO composite via HRTEM are qualitatively consistent with the 2-peak deconvolution of its corresponding XRD pattern, as shown in the middle panel. Still, a contribution of the sp
2 network to aromatic ring alignment cannot be ruled out.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}