
Yeast Nanobiotechnology



Abstract
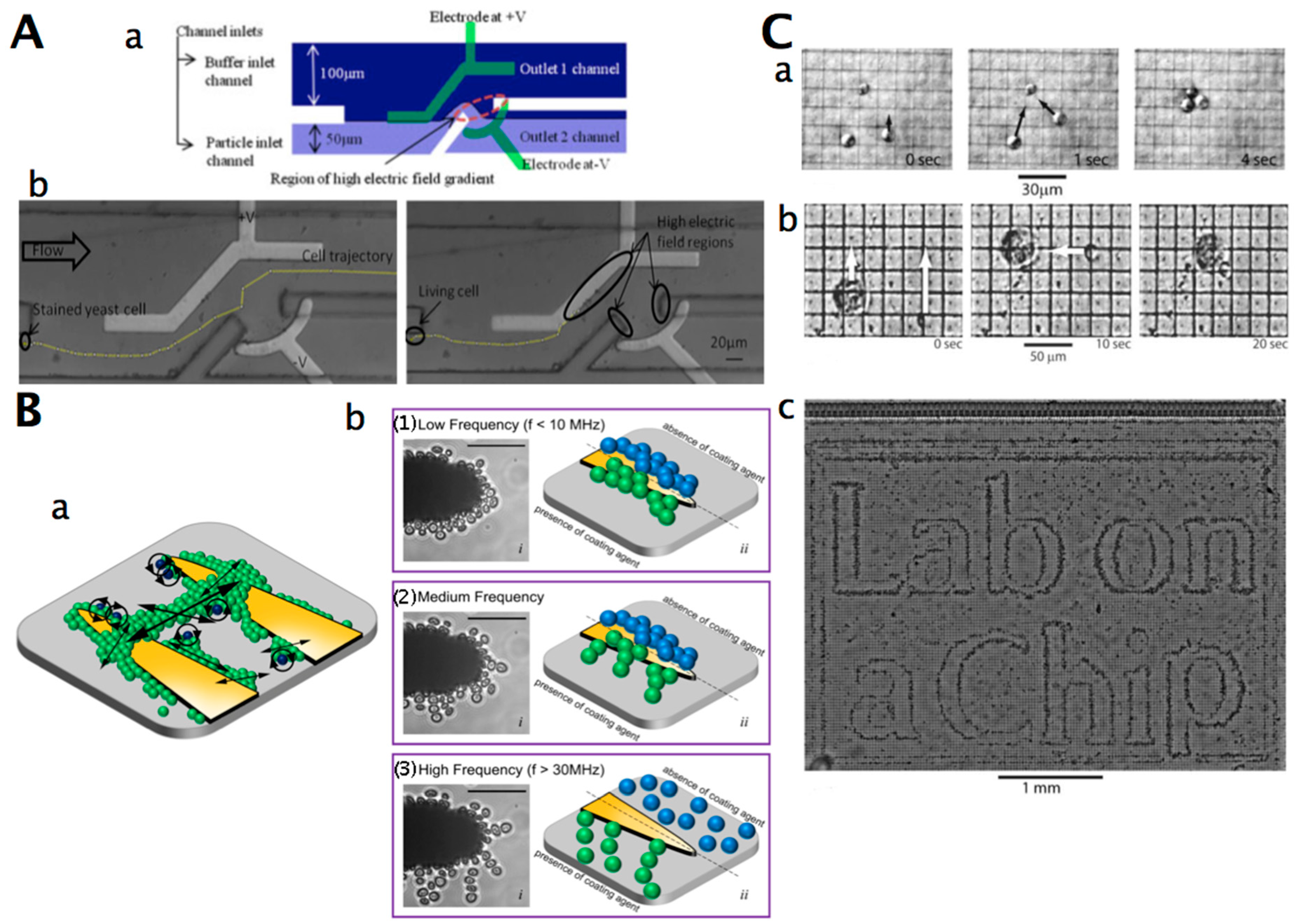
:1. Introduction
2. Yeast Nanobiotechnological Analyses
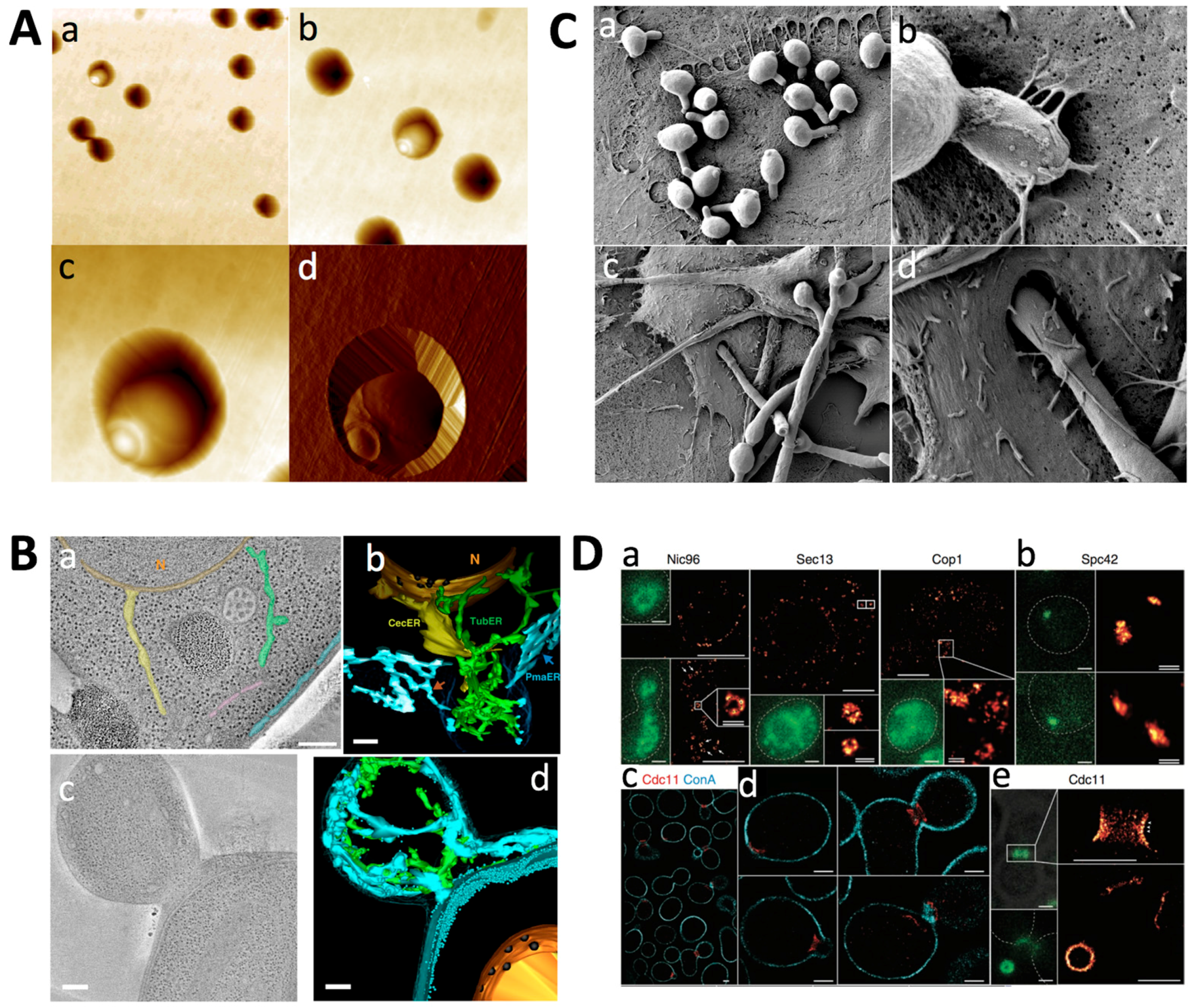
2.1. Nanoscale Imaging
2.1.1. Atomic Force Microscopy
2.1.2. Light Microscopy
2.1.3. Electron Microscopy
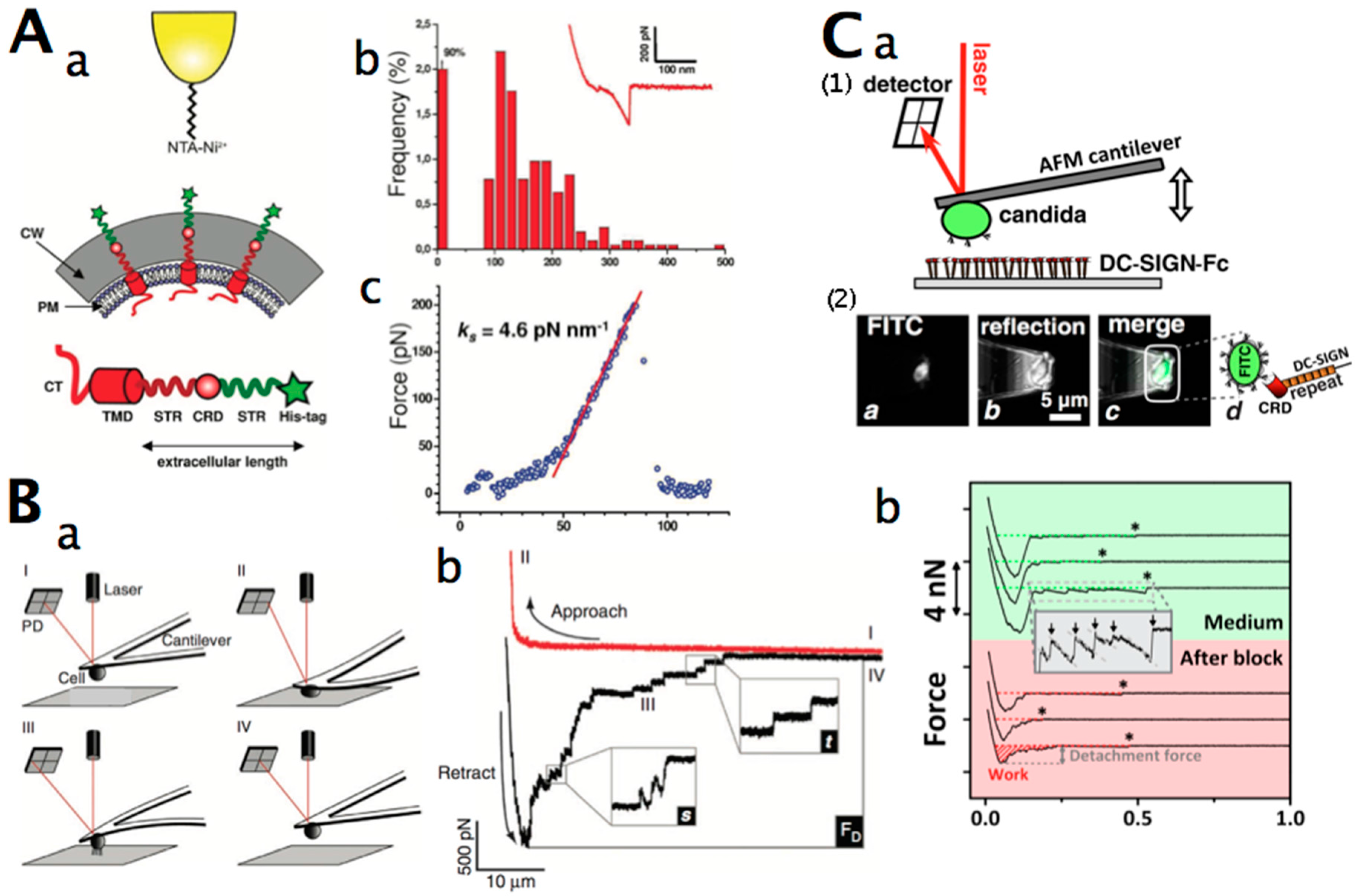
2.2. Force Microscopy
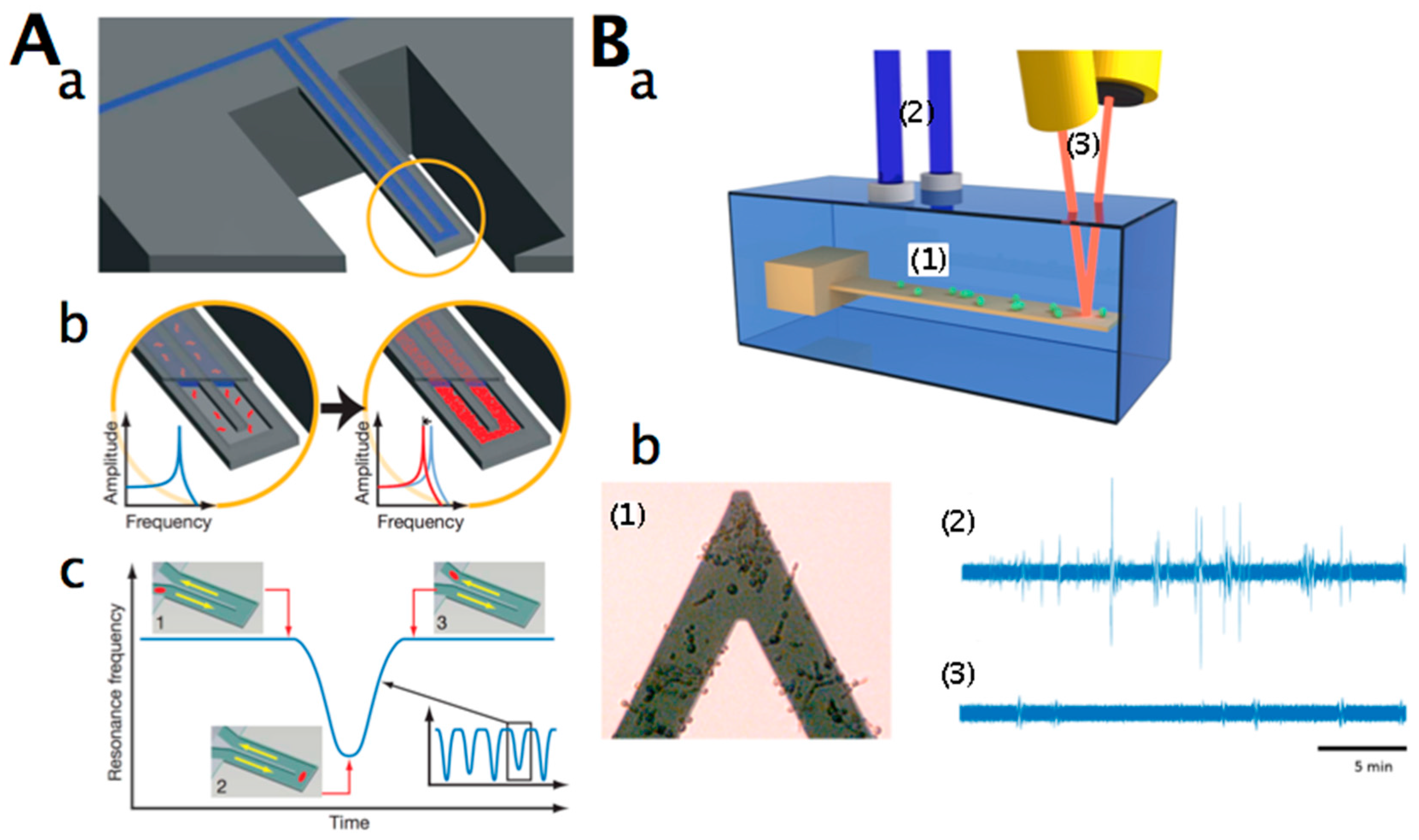
2.3. Nanomotion Analysis
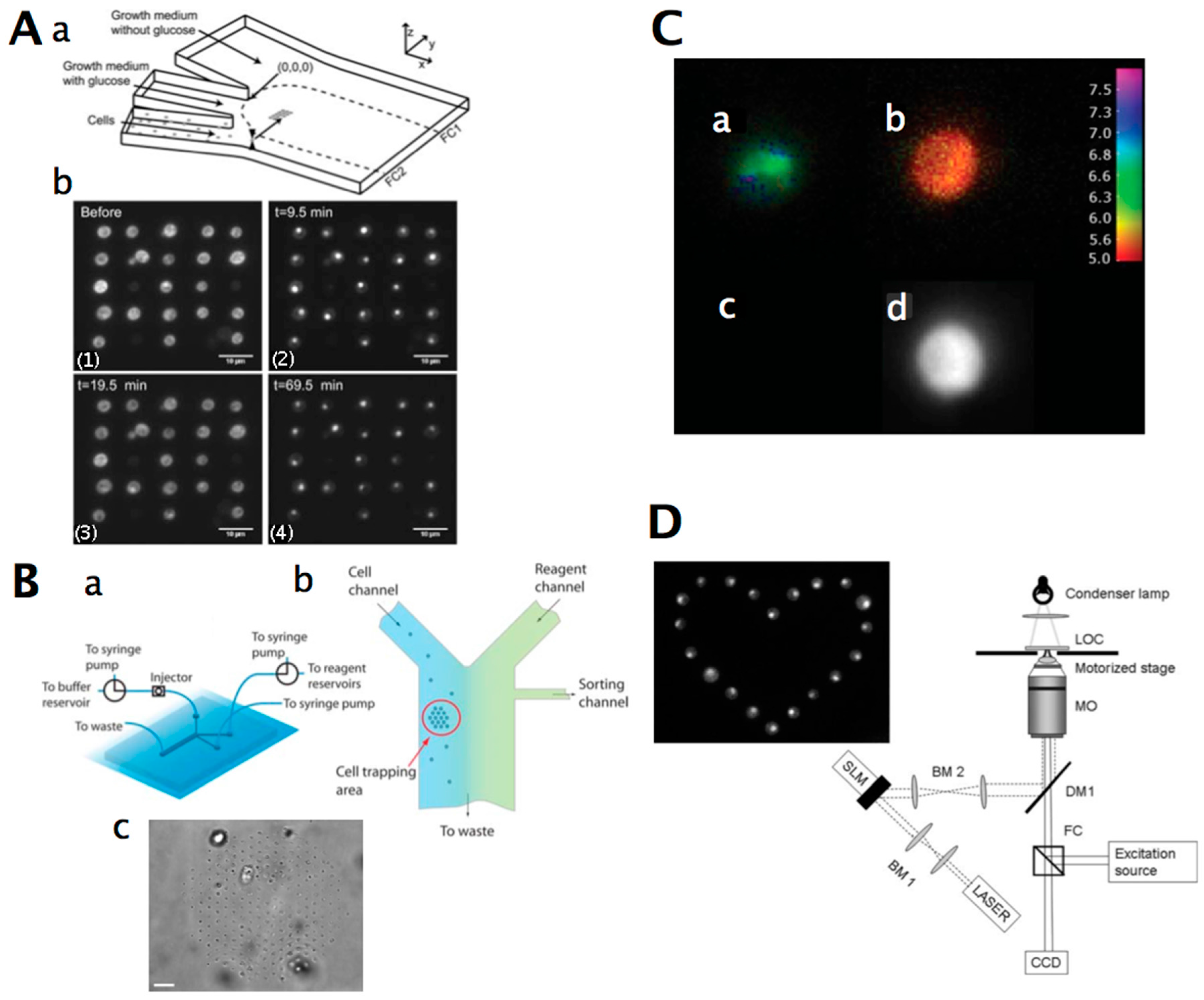
3. Yeast Cell Patterning and Manipulation
3.1. Yeast Cell Patterning
3.2. Direct Contact Cell Manipulation
3.2.1. AFM-Based Cell Manipulation
3.2.2. Micropipette Manipulation of Single Yeast Cells
3.3. Non-Contact Cell Manipulation
3.3.1. Optical Manipulation of Single Yeast Cells
3.3.2. Electrical and Magnetic Manipulation of Yeast Cells
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Roco, M.C.; Williams, R.S.; Alivisatos, P. (Eds.) Biological, Medical and Health Applications: Nanotechnology Research Directions; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000.
- Roco, M.C. Nanotechnology: Convergence with modern biology and medicine. Curr. Opin. Biotechnol. 2003, 14, 337–346. [Google Scholar] [CrossRef]
- Whitesides, G.M. The ‘right’ size in nanobiotechnology. Nat. Biotechnol. 2003, 21, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Binnig, G.; Rohrer, H. Scanning tunnelling microscopy. Helv. Phys. Acta 1982, 55, 726–735. [Google Scholar]
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic force microscope. Phys. Rev. Lett. 1986, 56, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Kada, G.; Kienberger, F.; Hinterdorfer, P. Atomic force microscopy in bionanotechnology. Nano Today 2008, 3, 12–19. [Google Scholar] [CrossRef]
- Ando, T. High-speed atomic force microscopy. Microscopy (Oxf.) 2013, 62, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Kasas, S.; Ikai, A. A method for anchoring round shaped cells for atomic force microscope imaging. Biophys. J. 1995, 68, 1678–1680. [Google Scholar] [CrossRef]
- Gad, M.; Ikai, A. Method for immobilizing microbial cells on gel surface for dynamic AFM studies. Biophys. J. 1995, 69, 2226–2233. [Google Scholar] [CrossRef]
- De, T.; Chettoor, A.M.; Agarwal, P.; Salapaka, M.V.; Nettikadan, S. Immobilization method of yeast cells for intermittent contact mode imaging using the atomic force microscope. Ultramicroscopy 2010, 110, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Dague, E.; Jauvert, E.; Laplatine, L.; Viallet, B.; Thibault, C.; Ressier, L. Assembly of live micro-organisms on microstructured PDMS stamps by convective/capillary deposition for AFM bio-experiments. Nanotechnology 2011, 22, 395102. [Google Scholar] [CrossRef] [PubMed]
- Formosa, C.; Pillet, F.; Schiavone, M.; Duval, R.E.; Ressier, L.; Dague, E. Generation of living cell arrays for atomic force microscopy studies. Nat. Protoc. 2015, 10, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Kasas, S.; Dietler, G. Probing nanomechanical properties from biomolecules to living cells. Pflugers Arch. 2008, 456, 13–27. [Google Scholar] [CrossRef] [PubMed]
- West, M.; Zurek, N.; Hoenger, A.; Voeltz, G.K. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J. Cell Biol. 2011, 193, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Ries, J.; Kaplan, C.; Platonova, E.; Eghlidi, H.; Ewers, H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods 2012, 9, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Formosa, C.; Schiavone, M.; Martin-Yken, H.; François, J.M.; Duval, R.E.; Dague, E. Nanoscale effects of caspofungin against two yeast species, Saccharomyces cerevisiae and Candida albicans. Antimicrob. Agents Chemother. 2013, 57, 3498–3506. [Google Scholar] [CrossRef] [PubMed]
- Chopinet, L.; Formosa, C.; Rols, M.P.; Duval, R.E.; Dague, E. Imaging living cells surface and quantifying its properties at high resolution using AFM in QI™ mode. Micron 2013, 48, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Formosa, C.; Schiavone, M.; Boisrame, A.; Richard, M.L.; Duval, R.E.; Dague, E. Multiparametric imaging of adhesive nanodomains at the surface of Candida albicans by atomic force microscopy. Nanomedicine 2015, 11, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Vilas, A.; Gallardo, A.M.; Perez-Giraldo, C.; Gonzalez-Martín, M.L.; Nuevo, M.J. Surface morphological characterization of yeast cells by scanning force microscopy. Surf. Interface Anal. 2001, 31, 1027–1030. [Google Scholar] [CrossRef]
- Gad, M.; Itoh, A.; Ikai, A. Mapping cell wall polysaccharides of living microbial cells using atomic force microscopy. Cell Biol. Int. 1997, 21, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Touhami, A.; Nysten, B.; Dufrêne, Y.F. Nanoscale mapping of the elasticity of microbial cells by Atomic Force Microscopy. Langmuir 2003, 19, 4539–4543. [Google Scholar] [CrossRef]
- Adya, A.K.; Canetta, E.; Walker, G.M. Atomic force microscopic study of the influence of physical stresses on Saccharomyces cerevisiae and Schizosaccharomyces pombe. FEMS Yeast Res. 2006, 6, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Pelling, A.E.; Sehati, S.; Gralla, E.B.; Gimzewski, J.K. Time dependence of the frequency and amplitude of the local nanomechanical motion of yeast. Nanomedicine 2005, 1, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Voychuk, S.I.; Gromozova, E.N.; Lytvyn, P.M.; Podgorsky, V.S. Changes of surface properties of yeast cell wall under exposure of electromagnetic field (40.68 MHz) and action of nystatin. Environmentalist 2005, 25, 139–144. [Google Scholar] [CrossRef]
- Stephens, D.J.; Allan, V.J. Light microscopy techniques for live cell imaging. Science 2003, 300, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Rajfur, Z.; Pomorski, P.; Jacobson, K. Microscope-based techniques to study cell adhesion and migration. Nat. Cell Biol. 2002, 4, E91–E96. [Google Scholar] [CrossRef] [PubMed]
- Gitai, Z. New fluorescence microscopy methods for microbiology: Sharper, faster, and quantitative. Curr. Opin. Microbiol. 2009, 12, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Runions, J.; Moore, I. Quantitative fluorescence microscopy: From art to science. Annu. Rev. Plant Biol. 2006, 57, 79–107. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Snapp, E.; Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2001, 2, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Jares-Erijman, E.A.; Jovin, T.M. FRET imaging. Nat. Biotechnol. 2003, 21, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Strutt, J.W. On the manufacture and theory of diffraction-gratings. Philos. Mag. 1874, 47, 193–205. [Google Scholar]
- Thompson, R.E.; Larson, D.R.; Webb, W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002, 82, 2775–2783. [Google Scholar] [CrossRef]
- Sako, Y. Imaging single molecules in living cells for systems biology. Mol. Syst. Biol. 2006, 2, 56. [Google Scholar] [CrossRef] [PubMed]
- Walter, N.G.; Huang, C.Y.; Manzo, A.J.; Sobhy, M.A. Do-it-yourself guide: How to use the modern single-molecule toolkit. Nat. Methods 2008, 5, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Dehmelt, L.; Bastiaens, P.I. Spatial organization of intracellular communication: Insights from imaging. Nat. Rev. Mol. Cell Biol. 2010, 11, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Harriss, L.M.; Wallace, M.I. Single molecule fluorescence in membrane biology. In Single Molecule Biology; Knight, A.E., Ed.; Academic Press: San Diego, CA, USA, 2009; pp. 253–288. [Google Scholar]
- Müller-Taubenberger, A.; Anderson, K.I. Recent advances using green and red fluorescent protein variants. Appl. Microbiol. Biotechnol. 2007, 77, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ando, R.; Hama, H.; Yamamoto-Hino, M.; Mizuno, H.; Miyawaki, A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 12651–12656. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Belousov, V.V.; Zaraisky, A.G.; Novoselov, V.V.; Staroverov, D.B.; Zorov, D.B.; Lukyanov, S.; Lukyanov, K.A. Kindling fluorescent proteins for precise in vivo photolabeling. Nat. Biotechnol. 2003, 21, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Patterson, G.H. Development and use of fluorescent protein markers in living cells. Science 2003, 300, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.M.; Verkhusha, V.V.; Staroverov, D.B.; Souslova, E.A.; Lukyanov, S.; Lukyanov, K.A. Photoswitchable cyan fluorescent protein for protein tracking. Nat. Biotechnol. 2004, 22, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Wiedenmann, J.; Ivanchenko, S.; Oswald, F.; Schmitt, F.; Röcker, C.; Salih, A.; Spindler, K.D.; Nienhaus, G.U. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15905–15910. [Google Scholar] [CrossRef] [PubMed]
- Gurskaya, N.G.; Verkhusha, V.V.; Shcheglov, A.S.; Staroverov, D.B.; Chepurnykh, T.V.; Fradkov, A.F.; Lukyanov, S.; Lukyanov, K.A. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol. 2006, 24, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Wiedenmann, J.; Nienhaus, G.U. Live-cell imaging with EosFP and other photoactivatable marker proteins of the GFP family. Expert Rev. Proteom. 2006, 3, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Patterson, G.H. Fluorescent proteins for photoactivation experiments. Methods Cell Biol. 2008, 85, 45–61. [Google Scholar] [PubMed]
- Patterson, G.H.; Lippincott-Schwartz, J. Selective photolabeling of proteins using photoactivatable GFP. Methods 2004, 32, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Habuchi, S.; Ando, R.; Dedecker, P.; Verheijen, W.; Mizuno, H.; Miyawaki, A.; Hofkens, J. Reversible single-molecule photoswitching in the GFP-like fluorescent protein Dronpa. Proc. Natl. Acad. Sci. USA 2005, 102, 9511–9516. [Google Scholar] [CrossRef] [PubMed]
- Verkhusha, V.V.; Sorkin, A. Conversion of the monomeric red fluorescent protein into a photoactivatable probe. Chem. Biol. 2005, 12, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; D’Angelo, C.; Oswald, F.; Denzel, A.; Mazel, C.H.; Matz, M.V.; Ivanchenko, S.; Nienhaus, G.U.; Wiedenmann, J. A green fluorescent protein with photoswitchable emission from the deep sea. PLoS ONE 2008, 3, e3766. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, D.; Adam, V. Reversible photoswitching in fluorescent proteins: A mechanistic view. IUBMB Life 2012, 64, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Lin, M.Z. Photoswitchable fluorescent proteins: Ten years of colorful chemistry and exciting applications. Curr. Opin. Chem. Biol. 2013, 17, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Adam, V.; Byrdin, M.; Bourgeois, D. Structural basis of photoswitching in fluorescent proteins. Methods Mol. Biol. 2014, 1148, 177–202. [Google Scholar] [PubMed]
- Nienhaus, G.U.; Nienhaus, K.; Hölzle, A.; Ivanchenko, S.; Renzi, F.; Oswald, F.; Wolff, M.; Schmitt, F.; Röcker, C.; Vallone, B.; et al. Photoconvertible fluorescent protein EosFP: Biophysical properties and cell biology applications. Photochem. Photobiol. 2006, 82, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Adam, V.; Nienhaus, K.; Bourgeois, D.; Nienhaus, G.U. Structural basis of enhanced photoconversion yield in green fluorescent protein-like protein Dendra2. Biochemistry 2009, 48, 4905–4915. [Google Scholar] [CrossRef] [PubMed]
- Lukyanov, K.A.; Chudakov, D.M.; Lukyanov, S.; Verkhusha, V.V. Innovation: Photoactivatable fluorescent proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gurskaya, N.G.; Merzlyak, E.M.; Staroverov, D.B.; Mudrik, N.N.; Samarkina, O.N.; Vinokurov, L.M.; Lukyanov, S.; Lukyanov, K.A. Method for real-time monitoring of protein degradation at the single cell level. Biotechniques 2007, 42, 446, 448, 450. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.H. Photoactivation and imaging of optical highlighter fluorescent proteins. Curr. Protoc. Cytom. 2011. [Google Scholar] [CrossRef]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.J.; Verkhusha, V.V.; Hess, S.T. Imaging biological structures with fluorescence photoactivation localization microscopy. Nat. Protoc. 2009, 4, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, D.; McEvoy, A.L.; Shroff, H.; Crooks, G.E.; Wingreen, N.S.; Betzig, E.; Liphardt, J. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. PLoS Biol. 2009, 7, e1000137. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Flors, C.; Hotta, J.; Uji-I, H.; Dedecker, P.; Ando, R.; Mizuno, H.; Miyawaki, A.; Hofkens, J. A stroboscopic approach for fast photoactivation-localization microscopy with Dronpa mutants. J. Am. Chem. Soc. 2007, 129, 13970–13977. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W. Far-field optical nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Stiel, A.C.; Andresen, M.; Bock, H.; Hilbert, M.; Schilde, J.; Schönle, A.; Eggeling, C.; Egner, A.; Hell, S.W.; Jakobs, S. Generation of monomeric reversibly switchable red fluorescent proteins for far-field fluorescence nanoscopy. Biophys. J. 2008, 95, 2989–2997. [Google Scholar] [CrossRef] [PubMed]
- Deschout, H.; Shivanandan, A.; Annibale, P.; Scarselli, M.; Radenovic, A. Progress in quantitative single-molecule localization microscopy. Histochem. Cell Biol. 2014, 142, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Juette, M.F.; Gould, T.J.; Lessard, M.D.; Mlodzianoski, M.J.; Nagpure, B.S.; Bennett, B.T.; Hess, S.T.; Bewersdorf, J. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 2008, 5, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.; Crossman, D.J. Technical review: Types of imaging-direct STORM. Anat. Rec. (Hoboken) 2014, 297, 2227–2231. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Merino, D. Stochastic optical reconstruction microscopy (STORM) in comparison with stimulated emission depletion (STED) and other imaging methods. J. Neurochem. 2015, 135, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.; Geisler, C.; Wurm, C.A.; von Middendorf, C.; Jakobs, S.; Schönle, A.; Egner, A.; Hell, S.W.; Eggeling, C. Two-color far-field fluorescence nanoscopy based on photoswitchable emitters. Appl. Phys. B 2007, 88, 161–165. [Google Scholar] [CrossRef]
- Egner, A.; Geisler, C.; von Middendorff, C.; Bock, H.; Wenzel, D.; Medda, R.; Andresen, M.; Stiel, A.C.; Jakobs, S.; Eggeling, C.; et al. Fluorescence nanoscopy in whole cells by asynchronous localization of photoswitching emitters. Biophys. J. 2007, 93, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed]
- Shroff, H.; Galbraith, C.G.; Galbraith, J.A.; White, H.; Gillette, J.; Olenych, S.; Davidson, M.W.; Betzig, E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc. Natl. Acad. Sci. USA 2007, 104, 20308–20313. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [PubMed]
- Donnert, G.; Keller, J.; Medda, R.; Andrei, M.A.; Rizzoli, S.O.; Lührmann, R.; Jahn, R.; Eggeling, C.; Hell, S.W. Macromolecular-scale resolution in biological fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2006, 103, 11440–11445. [Google Scholar] [CrossRef] [PubMed]
- Willig, K.I.; Rizzoli, S.O.; Westphal, V.; Jahn, R.; Hell, S.W. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature 2006, 440, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Westphal, V.; Rizzoli, S.O.; Lauterbach, M.A.; Kamin, D.; Jahn, R.; Hell, S.W. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science 2008, 320, 246–249. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, N.; Peckys, D.B.; Kremers, G.J.; Piston, D.W. Electron microscopy of whole cells in liquid with nanometer resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 2159–2164. [Google Scholar] [CrossRef] [PubMed]
- Peckys, D.B.; de Jonge, N. Liquid scanning transmission electron microscopy: Imaging protein complexes in their native environment in whole eukaryotic cells. Microsc. Microanal. 2014, 20, 346–365. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, W.; Grimm, R.; Walz, J. Electron tomography of molecules and cells. Trends Cell Biol. 1999, 9, 81–85. [Google Scholar] [CrossRef]
- Downing, K.H.; Sui, H.; Auer, M. Electron tomography: A 3D view of the subcellular world. Anal. Chem. 2007, 79, 7949–7957. [Google Scholar] [CrossRef] [PubMed]
- Stahlberg, H.; Walz, T. Molecular electron microscopy: State of the art and current challenges. ACS Chem. Biol. 2008, 3, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Lucić, V.; Leis, A.; Baumeister, W. Cryo-electron tomography of cells: Connecting structure and function. Histochem. Cell Biol. 2008, 130, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Diebolder, C.A.; Koster, A.J.; Koning, R.I. Pushing the resolution limits in cryo electron tomography of biological structures. J. Microsc. 2012, 248, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Agar, H.D.; Douglas, H.C. Studies on the cytological structure of yeast: Electron microscopy of thin sections. J. Bacteriol. 1957, 73, 365–375. [Google Scholar] [PubMed]
- Matile, P.; Moor, H.; Robinow, C.F. Yeast cytology. In The Yeasts; Rose, A.H., Harrison, J.S., Eds.; Avademic Press: New York, NY, USA, 1969; pp. 219–302. [Google Scholar]
- Osumi, M. Visualization of yeast cells by electron microscopy. J. Electron. Microsc. (Tokyo) 2012, 61, 343–365. [Google Scholar] [CrossRef] [PubMed]
- Osumi, M.; Sando, N. Division of yeast mitochondria in synchronous culture. J. Electron. Microsc. (Tokyo) 1969, 18, 47–56. [Google Scholar] [PubMed]
- Byers, B.; Goetsch, L. Duplication of spindle plaques and integration of the yeast cell cycle. Cold Spring Harb. Symp. Quant. Biol. 1974, 38, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Byers, B.; Goetsch, L. Behavior of spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. J. Bacteriol. 1975, 124, 511–523. [Google Scholar] [PubMed]
- Osumi, M.; Imaizumi, F.; Imai, M.; Sato, H.; Yamaguchi, H. Isolation and characterization of microbodies from candida tropicalis pk 233 cells grown on normal alkanes. J. Gen. Appl. Microbiol. 1975, 21, 375–387. [Google Scholar] [CrossRef]
- Novick, P.; Field, C.; Schekman, R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 1980, 21, 205–215. [Google Scholar] [CrossRef]
- Winey, M.; Goetsch, L.; Baum, P.; Byers, B. MPS1 and MPS2: Novel yeast genes defining distinct steps of spindle pole body duplication. J. Cell Biol. 1991, 114, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Osumi, M. Transmission and scanning electron microscopic examination of intracellular organelles in freeze-substituted Kloeckera and Saccharomyces cerevisiae yeast cells. J. Electron. Microsc. Tech. 1987, 5, 249–261. [Google Scholar] [CrossRef]
- Giddings, T.H., Jr.; O’Toole, E.T.; Morphew, M.; Mastronarde, D.N.; McIntosh, J.R.; Winey, M. Using rapid freeze and freeze-substitution for the preparation of yeast cells for electron microscopy and three-dimensional analysis. Methods Cell Biol. 2001, 67, 27–42. [Google Scholar] [PubMed]
- McDonald, K. Cryopreparation methods for electron microscopy of selected model systems. Methods Cell Biol. 2007, 79, 23–56. [Google Scholar] [PubMed]
- O’Toole, E.T.; Giddings, T.H., Jr.; Winey, M. Building cell structures in three dimensions: Electron tomography methods for budding yeast. In Yeast Protocols; CSHL Press: Cold Spring Harbor, NY, USA, 2016; pp. 303–312. [Google Scholar]
- Hoenger, A.; McIntosh, J.R. Probing the macromolecular organization of cells by electron tomography. Curr. Opin. Cell Biol. 2009, 21, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Arfsten, J.; Leupold, S.; Bradtmöller, C.; Kampen, I.; Kwade, A. Atomic force microscopy studies on the nanomechanical properties of Saccharomyces cerevisiae. Colloids Surf. B Biointerfaces 2010, 79, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Alsteens, D.; Beaussart, A.; Derclaye, S.; El-Kirat-Chatel, S.; Park, H.R.; Lipke, P.N.; Dufrêne, Y.F. Single-cell force spectroscopy of Als-mediated fungal adhesion. Anal. Methods 2013, 5, 3657–3662. [Google Scholar] [CrossRef] [PubMed]
- Portillo, A.M.; Krasnoslobodtsev, A.V.; Lyubchenko, Y.L. Effect of electrostatics on aggregation of prion protein Sup35 peptide. J. Phys. Condens. Matter 2012, 24, 164205. [Google Scholar] [CrossRef] [PubMed]
- Portillo, A.; Hashemi, M.; Zhang, Y.; Breydo, L.; Uversky, V.N.; Lyubchenko, Y.L. Role of monomer arrangement in the amyloid self-assembly. Biochim. Biophys. Acta 2015, 1854, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Rangl, M.; Ebner, A.; Yamada, J.; Rankl, C.; Tampé, R.; Gruber, H.J.; Rexach, M.; Hinterdorfer, P. Single-molecule analysis of the recognition forces underlying nucleo-cytoplasmic transport. Angew. Chem. Int. Ed. Engl. 2013, 52, 10356–10359. [Google Scholar] [CrossRef] [PubMed]
- Goossens, K.V.Y.; Ielasi, F.S.; Nookaew, I.; Stals, I.; Alonso-Sarduy, L.; Daenen, L.; van Mulders, S.E.; Stassen, C.; van Eijsden, R.G.E.; Siewers, V.; et al. Molecular mechanism of flocculation self-recognition in yeast and its role in mating and survival. mBio 2015, 6, e00427–e00415. [Google Scholar] [CrossRef] [PubMed]
- El-Kirat-Chatel, S.; Beaussart, A.; Alsteens, D.; Sarazin, A.; Jouault, T.; Dufrêne, Y.F. Single-molecule analysis of the major glycopolymers of pathogenic and non-pathogenic yeast cells. Nanoscale 2013, 5, 4855–4863. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.; Marsh, G.; Gao, L.; Waugh, R.; Koo, H. Binding force dynamics of Streptococcus mutans-glucosyltransferase B to Candida albicans. J. Dent. Res. 2015, 94, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- El-Kirat-Chatel, S.; Beaussart, A.; Derclaye, S.; Alsteens, D.; Kucharíková, S.; van Dijck, P.; Dufrêne, Y.F. Force nanoscopy of hydrophobic interactions in the fungal pathogen Candida glabrata. ACS Nano 2015, 9, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Touhami, A.; Hoffmann, B.; Vasella, A.; Denis, F.A.; Dufrêne, Y.F. Aggregation of yeast cells: Direct measurement of discrete lectin-carbohydrate interactions. Microbiology 2003, 149, 2873–2878. [Google Scholar] [CrossRef] [PubMed]
- Dupres, V.; Alsteens, D.; Wilk, S.; Hansen, B.; Heinisch, J.J.; Dufrêne, Y.F. The yeast Wsc1 cell surface sensor behaves like a nanospring in vivo. Nat. Chem. Biol. 2009, 5, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Dupres, V.; Heinisch, J.J.; Dufrêne, Y.F. Atomic force microscopy demonstrates that disulphide bridges are required for clustering of the yeast cell wall integrity sensor Wsc1. Langmuir 2011, 27, 15129–15134. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, J.J.; Dupres, V.; Wilk, S.; Jendretzki, A.; Dufrêne, Y.F. Single-molecule atomic force microscopy reveals clustering of the yeast plasma-membrane sensor Wsc1. PLoS ONE 2010, 5, e11104. [Google Scholar] [CrossRef] [PubMed]
- Formosa, C.; Lachaize, V.; Galés, C.; Rols, M.P.; Martin-Yken, H.; François, J.M.; Duval, R.E.; Dague, E. Mapping HA-tagged protein at the surface of living cells by atomic force microscopy. J. Mol. Recognit. 2015, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.; Miyachi, Y.; Ishii, J.; Ogino, C.; Kondo, A. The mapping of yeast’s G-protein coupled receptor with an atomic force microscope. Nanoscale 2015, 7, 4956–4963. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, J.; Helenius, J.; Muller, D.J. Quantifying cellular adhesion to extracellular matrix components by single-cell force spectroscopy. Nat. Protoc. 2010, 5, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Te Riet, J.; Reinieren-Beeren, I.; Figdor, C.G.; Cambi, A. AFM force spectroscopy reveals how subtle structural differences affect the interaction strength between Candida albicans and DC-SIGN. J. Mol. Recognit. 2015, 28, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Gabriel, D.; Gerisch, G.; Gaub, H.E. Discrete interactions in cell adhesion measured by single-molecule force spectroscopy. Nat. Cell Biol. 2000, 2, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Helenius, J.; Heisenberg, C.P.; Gaub, H.E.; Muller, D.J. Single-cell force spectroscopy. J. Cell Sci. 2008, 121, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, J.; Legate, K.R.; Schubert, R.; Bharadwaj, M.; Werner, C.; Müller, D.J.; Benoit, M. A practical guide to quantify cell adhesion using single-cell force spectroscopy. Methods 2013, 60, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, A.; Cisneros, D.A.; Friedrichs, J.; Puech, P.H.; Muller, D.J.; Franz, C.M. Revealing early steps of alpha2beta1 integrin-mediated adhesion to collagen type I by using single-cell force spectroscopy. Mol. Biol. Cell 2007, 18, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.M.; Ovchinnikova, E.S.; Krom, B.P.; Schlecht, L.M.; Zhou, H.; Hoyer, L.L.; Busscher, H.J.; van der Mei, H.C.; Jabra-Rizk, M.A.; Shirtliff, M.E. Staphylococcus aureus adherence to Candida albicans hyphae is mediated by the hyphal adhesin Als3p. Microbiology 2012, 158, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, E.; Guillaume-Gentil, O.; Ossola, D.; Polesel-Maris, J.; LeibundGut-Landmann, S.; Zambelli, T.; Vorholt, J.A. Rapid and serial quantification of adhesion forces of yeast and Mammalian cells. PLoS ONE 2012, 7, e52712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsteens, D.; van Dijck, P.; Lipke, P.N.; Dufrêne, Y.F. Quantifying the forces driving cell-cell adhesion in a fungal pathogen. Langmuir 2013, 29, 13473–13480. [Google Scholar] [CrossRef] [PubMed]
- Bowen, W.R.; Lovitt, R.W.; Wright, C.J. Atomic Force Microscopy study of the adhesion of Saccharomyces cerevisiae. J. Colloid Interface Sci. 2001, 237, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Götzinger, M.; Weigl, B.; Peukert, W.; Sommer, K. Effect of roughness on particle adhesion in aqueous solutions: A study of Saccharomyces cerevisiae and a silica particle. Colloids Surf. B Biointerfaces 2007, 55, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Hachułka, K.; Lekka, M.; Okrajni, J.; Ambroziak, W.; Wandelt, B. Polymeric sensing system molecularly imprinted towards enhanced adhesion of Saccharomyces cerevisiae. Biosens. Bioelectron. 2010, 26, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.; Behr, P.; Drechsler, U.; Polesel-Maris, J.; Potthoff, E.; Vörös, J.; Zambelli, T. SU-8 hollow cantilevers for AFM cell adhesion studies. J. Micromech. Microeng. 2016, 26, 055006. [Google Scholar] [CrossRef]
- Hansen, K.M.; Thundat, T. Microcantilever biosensors. Methods 2005, 37, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J. Cantilever biosensors. Analyst 2008, 133, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Ghatkesar, M.K.; Backmann, N.; Grange, W.; Boulanger, P.; Letellier, L.; Lang, H.P.; Bietsch, A.; Gerber, C.; Hegner, M. Quantitative time-resolved measurement of membrane protein-ligand interactions using microcantilever array sensors. Nat. Nanotechnol. 2009, 4, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Ndieyira, J.W.; Watari, M.; Barrera, A.D.; Zhou, D.; Vögtli, M.; Batchelor, M.; Cooper, M.A.; Strunz, T.; Horton, M.A.; Abell, C.; et al. Nanomechanical detection of antibiotic-mucopeptide binding in a model for superbug drug resistance. Nat. Nanotechnol. 2008, 3, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Godin, M.; Delgado, F.F.; Son, S.; Grover, W.H.; Bryan, A.K.; Tzur, A.; Jorgensen, P.; Payer, K.; Grossman, A.D.; Kirschner, M.W.; et al. Using buoyant mass to measure the growth of single cells. Nat. Methods 2010, 7, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.P.; Baller, M.K.; Berger, R.; Gerber, C.; Gimzewski, J.K.; Battiston, F.M.; Fornaro, P.; Ramseyer, J.P.; Meyer, E.; Guntherodt, H.J. An artificial nose based on a micromechancial cantilever array. Anal. Chim. Acta 1999, 393, 59–65. [Google Scholar] [CrossRef]
- Braun, T.; Barwich, V.; Ghatkesar, M.K.; Bredekamp, A.H.; Gerber, C.; Hegner, M.; Lang, H.P. Micromechanical mass sensors for biomolecular detection in a physiological environment. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2005, 72, 031907. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, S.; Chiyoma, T.; Ikeuchi, A.; Okano, H.; Sone, H.; Izumi, T. Possibility of a femtogram mass biosensor using a self-sensing cantilever. Curr. Appl. Phys. 2006, 6, 384–388. [Google Scholar] [CrossRef]
- Liu, Y.; Schweizerb, L.M.; Wanga, W.; Reubena, R.L.; Schweizer, M.; Shu, W. Label-free and real-time monitoring of yeast cell growth by the bending of polymer microcantilever biosensors. Sens. Actuator B Chem. 2013, 178, 621–626. [Google Scholar] [CrossRef]
- Bryan, A.K.; Goranov, A.; Amon, A.; Manalis, S.R. Measurement of mass, density, and volume during the cell cycle of yeast. Proc. Natl. Acad. Sci. USA 2010, 107, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Godin, M.; Tabard-Cossa, V.; Miyahara, Y.; Monga, T.; Williams, P.J.; Beaulieu, L.Y.; Bruce Lennox, R.; Grutter, P. Cantilever-based sensing: The origin of surface stress and optimization strategies. Nanotechnology 2010, 21, 75501. [Google Scholar] [CrossRef] [PubMed]
- Burg, T.P.; Godin, M.; Knudsen, S.M.; Shen, W.; Carlson, G.; Foster, J.S.; Babcock, K.; Manalis, S.R. Weighing of biomolecules, single cells and single nanoparticles in fluid. Nature 2007, 446, 1066–1069. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Jang, J.; Irimia, D.; Sturgis, J.; Lee, J.; Robinson, J.P.; Toner, M.; Bashir, R. ‘Living cantilever arrays’ for characterization of mass of single live cells in fluids. Lab Chip 2008, 8, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.K.; Hecht, V.C.; Shen, W.; Payer, K.; Grover, W.H.; Manalis, S.R. Measuring single cell mass, volume, and density with dual suspended microchannel resonators. Lab Chip 2014, 14, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Nugaeva, N.; Gfeller, K.Y.; Backmann, N.; Lang, H.P.; Düggelin, M.; Hegner, M. Micromechanical cantilever array sensors for selective fungal immobilization and fast growth detection. Biosens. Bioelectron. 2005, 21, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Aghayee, S.; Benadiba, C.; Notz, J.; Kasas, S.; Dietler, G.; Longo, G. Combination of fluorescence microscopy and nanomotion detection to characterize bacteria. J. Mol. Recognit. 2013, 26, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Longo, G.; Alonso-Sarduy, L.; Rio, L.M.; Bizzini, A.; Trampuz, A.; Notz, J.; Dietler, G.; Kasas, S. Rapid detection of bacterial resistance to antibiotics using AFM cantilevers as nanomechanical sensors. Nat. Nanotechnol. 2013, 8, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Kasas, S.; Ruggeri, F.S.; Benadiba, C.; Maillard, C.; Stupar, P.; Tournu, H.; Dietler, G.; Longo, G. Detecting nanoscale vibrations as signature of life. Proc. Natl. Acad. Sci. USA 2015, 112, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Voldman, J. Engineered systems for the physical manipulation of single cells. Curr. Opin. Biotechnol. 2006, 17, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Voldman, J. Electrical forces for microscale cell manipulation. Annu. Rev. Biomed. Eng. 2006, 8, 425–454. [Google Scholar] [CrossRef] [PubMed]
- Willaert, R.G.; Goossens, K. Microfluidic bioreactors for cellular microarrays. Fermentation 2015, 1, 38–78. [Google Scholar] [CrossRef]
- Guo, L.J. Nanoimprint lithography: Methods and material requirements. Adv. Mater. 2007, 19, 495–513. [Google Scholar] [CrossRef]
- Yap, F.L.; Zhang, Y. Protein and cell micropatterning and its integration with micro/nanoparticles assembly. Biosens. Bioelectron. 2007, 22, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Anselme, K.; Davidson, P.; Popa, A.M.; Giazzon, M.; Liley, M.; Ploux, L. The interaction of cells and bacteria with surfaces structured at the nanometre scale. Acta Biomater. 2010, 6, 3824–3846. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Xia, Y.; Whitesides, G.M. Soft lithography for micro- and nanoscale patterning. Nat. Protoc. 2010, 5, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Ekerdt, B.L.; Segalman, R.A.; Schaffer, D.V. Spatial organization of cell-adhesive ligands for advanced cell culture. Biotechnol. J. 2013, 8, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.; Patil, R.; Thombre, D.K.; Gade, W.N. Micro-nanopatterning as tool to study the role of physicochemical properties on cell-surface interactions. J. Biomed. Mater. Res. A 2013, 101, 3019–3032. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.H.; Ball, G.I.; McQuaide, S.C.; Chao, S.; Colman-Lerner, A.; Holl, M.R.; Meldrum, D.R. Microprinting of on-chip cultures: Patterning of yeast cell microarrays using concanavalin-A adhesion. In Proceedings of the IMECE04 ASME International Mechanical Engineering Congress, Anaheim, CA, USA, 13–19 November 2004; pp. 373–374.
- Cookson, S.; Ostroff, N.; Pang, W.L.; Volfson, D.; Hasty, J. Monitoring dynamics of single-cell gene expression over multiple cell cycles. Mol. Syst. Biol. 2005, 1, 0024. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, R.; Niu, W.; Scouras, A.D.; Hart, G.T.; Davies, J.; Ellington, A.D.; Iyer, V.R.; Marcotte, E.M. Systematic profiling of cellular phenotypes with spotted cell microarrays reveals mating-pheromone response genes. Genome Biol. 2006, 7, R6. [Google Scholar] [CrossRef] [PubMed]
- Ryley, J.; Pereira-Smith, O.M. Microfluidics device for single cell gene expression analysis in Saccharomyces cerevisiae. Yeast 2006, 23, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, R.; Moradi, E.K.; Niu, W.; Hart, G.T.; Davis, M.; McGary, K.L.; Ellington, A.D.; Marcotte, E.M. Systematic definition of protein constituents along the major polarization axis reveals an adaptive reuse of the polarization machinery in pheromone-treated budding yeast. J. Proteom. Res. 2009, 8, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Falconnet, D.; Niemistö, A.; Taylor, R.J.; Ricicova, M.; Galitski, T.; Shmulevich, I.; Hansen, C.L. High-throughput tracking of single yeast cells in a microfluidic imaging matrix. Lab Chip 2011, 11, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.; Eyer, K.; Robinson, T.; Schmidt, F.I.; Mercer, J.; Dittrich, P.S. A facile protocol for the immobilisation of vesicles, virus particles, bacteria, and yeast cells. Integr. Biol. (Camb.) 2012, 4, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Avalos Vizcarra, I.; Huberts, D.H.; Lee, L.P.; Heinemann, M. Whole lifespan microscopic observation of budding yeast aging through a microfluidic dissection platform. Proc. Natl. Acad. Sci. USA 2012, 109, 4916–4920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.C.; Hur, J.Y.; Cho, H.S.; Park, S.H.; Suh, K.Y. High-throughput single-cell quantification using simple microwell-based cell docking and programmable time-course live-cell imaging. Lab Chip 2011, 11, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, Y.; Zou, K.; Brandman, O.; Luo, C.; Ouyang, Q.; Li, H. Molecular phenotyping of aging in single yeast cells using a novel microfluidic device. Aging Cell 2012, 11, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, C.; Zou, K.; Xie, Z.; Brandman, O.; Ouyang, Q.; Li, H. Single cell analysis of yeast replicative aging using a new generation of microfluidic device. PLoS ONE 2012, 7, e48275. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, S.; Paoletti, C.; Goulev, Y.; Ungureanu, A.; Aguilaniu, H.; Charvin, G. Aging yeast cells undergo a sharp entry into senescence unrelated to the loss of mitochondrial membrane potential. Cell Rep. 2013, 5, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Crane, M.M.; Clark, I.B.; Bakker, E.; Smith, S.; Swain, P.S. A microfluidic system for studying ageing and dynamic single-cell responses in budding yeast. PLoS ONE 2014, 9, e100042. [Google Scholar] [CrossRef] [PubMed]
- Osada, K.; Hosokawa, M.; Yoshino, T.; Tanaka, T. Monitoring of cellular behaviors by microcavity array-based single-cell patterning. Analyst 2014, 139, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Young, T.Z.; Acar, M. Yeast replicator: A high-throughput multiplexed microfluidics platform for automated measurements of single-cell aging. Cell Rep. 2015, 13, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.C.; Liu, W.; Gu, L.; Dang, W.; Qin, L. High-throughput analysis of yeast replicative aging using a microfluidic system. Proc. Natl. Acad. Sci. USA 2015, 112, 9364–9369. [Google Scholar] [CrossRef] [PubMed]
- Terenna, C.R.; Makushok, T.; Velve-Casquillas, G.; Baigl, D.; Chen, Y.; Bornens, M.; Paoletti, A.; Piel, M.; Tran, P.T. Physical mechanisms redirecting cell polarity and cell shape in fission yeast. Curr. Biol. 2008, 18, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Minc, N.; Boudaoud, A.; Chang, F. Mechanical forces of fission yeast growth. Curr. Biol. 2009, 19, 1096–1101, Erratum in: Curr. Biol. 2014, 24, 1436. [Google Scholar] [CrossRef] [PubMed]
- Nghe, P.; Boulineau, S.; Gude, S.; Recouvreux, P.; van Zon, J.S.; Tans, S.J. Microfabricated polyacrylamide devices for the controlled culture of growing cells and developing organisms. PLoS ONE 2013, 8, e75537. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Luo, C.; Ouyang, Q. A microfluidic synchronizer for fission yeast cells. Lab Chip 2013, 13, 4071–4077. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.; Seshia, A.; Lando, D.; Laue, E.; Palayret, M.; Lee, S.F.; Klenerman, D. A microfluidic device for the hydrodynamic immobilisation of living fission yeast cells for super-resolution imaging. Sens. Actuators B Chem. 2014, 192, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Nobs, J.B.; Maerkl, S.J. Long-term single cell analysis of S. pombe on a microfluidic microchemostat array. PLoS ONE 2014, 9, e93466. [Google Scholar] [CrossRef] [PubMed]
- Spivey, E.C.; Xhemalce, B.; Shear, J.B.; Finkelstein, I.J. 3D-printed microfluidic microdissector for high-throughput studies of cellular aging. Anal. Chem. 2014, 86, 7406–7412. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.A.; Chen, C.S. Microcontact printing: A tool to pattern. Soft Matter 2007, 3, 168–177. [Google Scholar] [CrossRef]
- Miermont, A.; Waharte, F.; Hu, S.; McClean, M.N.; Bottani, S.; Léon, S.; Hersen, P. Severe osmotic compression triggers a slowdown of intracellular signaling, which can be explained by molecular crowding. Proc. Natl. Acad. Sci. USA 2013, 110, 5725–5730. [Google Scholar] [CrossRef] [PubMed]
- Théry, M.; Piel, M. Adhesive micropatterns for cells: A microcontact printing protocol. Cold Spring Harb. Protoc. 2009, 7. [Google Scholar] [CrossRef] [PubMed]
- Weaver, W.M.; Tseng, P.; Kunze, A.; Masaeli, M.; Chung, A.J.; Dudani, J.S.; Kittur, H.; Kulkarni, R.P.; di Carlo, D. Advances in high-throughput single-cell microtechnologies. Curr. Opin. Biotechnol. 2014, 25, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Fritzsch, F.S.; Dusny, C.; Frick, O.; Schmid, A. Single-cell analysis in biotechnology, systems biology, and biocatalysis. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 129–155. [Google Scholar] [CrossRef] [PubMed]
- Dusny, C.; Schmid, A. Microfluidic single-cell analysis links boundary environments and individual microbial phenotypes. Environ. Microbiol. 2015, 17, 1839–1856. [Google Scholar] [CrossRef] [PubMed]
- Groisman, A.; Lobo, C.; Cho, H.; Campbell, J.K.; Dufour, Y.S.; Stevens, A.M.; Levchenko, A. A microfluidic chemostat for experiments with bacterial and yeast cells. Nat. Methods 2005, 2, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Nugent, E.; Javer, A.; Cicuta, P.; Sclavi, B.; Cosentino Lagomarsino, M.; Dorfman, K.D. Microfluidic chemostat for measuring single cell dynamics in bacteria. Lab Chip 2013, 13, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Pang, W.L.; Ostroff, N.A.; Baumgartner, B.L.; Nayak, S.; Tsimring, L.S.; Hasty, J. Metabolic gene regulation in a dynamically changing environment. Nature 2008, 454, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Yarmush, M.L.; King, K.R. Living-cell microarrays. Annu. Rev. Biomed. Eng. 2009, 11, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Evander, M.; Hammarström, B.; Laurell, T. Review of cell and particle trapping in microfluidic systems. Anal. Chim. Acta 2009, 649, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, D.; Wu, L.Y.; Lee, L.P. Dynamic single cell culture array. Lab Chip 2006, 6, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Barbulovic-Nad, I.; Lucente, M.; Sun, Y.; Zhang, M.; Wheeler, A.R.; Bussmann, M. Bio-microarray fabrication techniques—A review. Crit. Rev. Biotechnol. 2006, 26, 237–259. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, F.; Souquet, A.; Catros, S.; Guillotin, B.; Lopez, J.; Faucon, M.; Pippenger, B.; Bareille, R.; Rémy, M.; Bellance, S.; et al. High-throughput laser printing of cells and biomaterials for tissue engineering. Acta Biomater. 2010, 6, 2494–2500. [Google Scholar] [CrossRef] [PubMed]
- Bean, G.J.; Jaeger, P.A.; Bahr, S.; Ideker, T. Development of ultra-high-density screening tools for microbial “omic”. PLoS ONE 2014, 9, e85177. [Google Scholar] [CrossRef] [PubMed]
- Schaack, B.; Reboud, J.; Combe, S.; Fouqué, B.; Berger, F.; Boccard, S.; Filhol-Cochet, O.; Chatelain, F. A “DropChip” cell array for DNA and siRNA transfection combined with drug screening. NanoBiotechnology 2005, 1, 183–189. [Google Scholar] [CrossRef]
- Ringeisen, B.R.; Othon, C.M.; Barron, J.A.; Young, D.; Spargo, B.J. Jet-based methods to print living cells. Biotechnol. J. 2006, 1, 930–948. [Google Scholar] [CrossRef] [PubMed]
- Roth, E.A.; Xu, T.; Das, M.; Gregory, C.; Hickman, J.J.; Boland, T. Inkjet printing for high-throughput cell patterning. Biomaterials 2004, 25, 3707–3715. [Google Scholar] [CrossRef] [PubMed]
- Ferris, C.J.; Gilmore, K.G.; Wallace, G.G.; In Het Panhuis, M. Biofabrication: An overview of the approaches used for printing of living cells. Appl. Microbiol. Biotechnol. 2013, 97, 4243–4258. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Macia, L.; Morrin, A.; Smyth, M.R.; Killard, A.J. Advanced printing and deposition methodologies for the fabrication of biosensors and biodevices. Analyst 2010, 135, 845–867. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rossignol, F.; Macdonald, J. Inkjet printing for biosensor fabrication: Combining chemistry and technology for advanced manufacturing. Lab Chip 2015, 15, 2538–2558. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Liley, M.; Brugger, J.; Pugin, R.; Heinzelmann, H. Nanodispenser for attoliter volume deposition using atomic force microscopy probes modified by focused-ion-beam milling. Appl. Phys. Lett. 2005, 85, 6260–6262. [Google Scholar] [CrossRef]
- Deladi, S.; Tas, N.R.; Berenschot, J.W.; de Boer, J.H.; de Boer, M.J.; Peter, M.; Krijnen, G.J.M.; Elwenspoek, M.C. Micromachines fountain pen for atomic force microscope-based nanopatterning. Appl. Phys. Lett. 2004, 85, 5361. [Google Scholar] [CrossRef]
- Kim, K.H.; Moldovan, N.; Espinosa, H.D. A nanofountain probe with sub-100 nm molecular writing resolution. Small 2005, 1, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Kawashima, T.; Shibata, T.; Mineta, T.; Makino, E. Micromachining of a newly designed AFM probe integrated with hollow microneedle for cellular function analysis. Microelectron. Eng. 2010, 87, 1185–1189. [Google Scholar] [CrossRef]
- Meister, A.; Gabi, M.; Behr, P.; Studer, P.; Vörös, J.; Niedermann, P.; Bitterli, J.; Polesel-Maris, J.; Liley, M.; Heinzelmann, H.; et al. FluidFM: Combining atomic force microscopy and nanofluidics in a universal liquid delivery system for single cell applications and beyond. Nano Lett. 2009, 9, 2501–2507. [Google Scholar] [CrossRef] [PubMed]
- Guillaume-Gentil, O.; Potthoff, E.; Ossola, D.; Franz, C.M.; Zambelli, T.; Vorholt, J.A. Force-controlled manipulation of single cells: From AFM to FluidFM. Trends Biotechnol. 2014, 32, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Dörig, P.; Stiefel, P.; Behr, P.; Sarajlic, E.; Bijl, D.; Gabi, M.; Vörös, J.; Vorholt, J.A.; Zambelli, T. Force-controlled spatial manipulation of viable mammalian cells and micro-organisms by means of FluidFM technology. Appl. Phys. Lett. 2010, 97, 023701. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.; Forró, C.; Weydert, S.; Aebersold, M.J.; Dermutz, H.; Guillaume-Gentil, O.; Zambelli, T.; Vörös, J.; Demkó, L. Controlled single-cell deposition and patterning by highly flexible hollow cantilevers. Lab Chip 2016, 16, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Sanford, K.K.; Earle, W.R.; Likely, G.D. The growth in vitro of single isolated tissue cells. J. Nat. Cancer Inst. 1948, 9, 229–246. [Google Scholar] [PubMed]
- Anis, Y.H.; Holl, M.R.; Meldrum, D.R. Automated selection and placement of single cells using vision-based feedback control. IEEE Trans. Autom. Sci. Eng. 2010, 7, 598. [Google Scholar] [CrossRef]
- Fröhlich, J.; König, H. New techniques for isolation of single prokaryotic cells. FEMS Microbiol. Rev. 2000, 24, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, S.; Xiao, J.; Silva, B.B.; Gonzalez, I.; Agidi, P.S.; Klein, M.I.; Ambatipudi, K.S.; Rosalen, P.L.; Bauserman, R.; Waugh, R.E.; et al. Role of glucosyltransferase B in interactions of Candida albicans with Streptococcus mutans and with an experimental pellicle on hydroxyapatite surfaces. Appl. Environ. Microbiol. 2011, 77, 6357–6367. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Moraes, C.; Ye, G.; Simmons, C.A.; Sun, Y. Single cell deposition and patterning with a robotic system. PLoS ONE 2010, 5, e13542. [Google Scholar] [CrossRef] [PubMed]
- Környei, Z.; Beke, S.; Mihálffy, T.; Jelitai, M.; Kovács, K.J.; Szabó, Z.; Szabó, B. Cell sorting in a Petri dish controlled by computer vision. Sci. Rep. 2013, 3, 1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungai-Salánki, R.; Gerecsei, T.; Fürjes, P.; Orgovan, N.; Sándor, N.; Holczer, E.; Horvath, R.; Szabó, B. Automated single cell isolation from suspension with computer vision. Sci. Rep. 2016, 6, 20375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roder, P.; Hille, C. A Multifunctional frontloading approach for repeated recycling of a pressure-controlled AFM micropipette. PLoS ONE 2015, 10, e0144157. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jeong, W.; Beebe, D.J. Microfluidic valve with cored glass microneedle for microinjection. Lab Chip 2003, 3, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.G.; Lin, F.; Jeon, N.L. A microfluidic multi-injector for gradient generation. Lab Chip 2006, 6, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, P.; Schmidt, F.I.; Dörig, P.; Behr, P.; Zambelli, T.; Vorholt, J.A.; Mercer, J. Cooperative vaccinia infection demonstrated at the single-cell level using FluidFM. Nano Lett. 2012, 12, 4219–4227. [Google Scholar] [CrossRef] [PubMed]
- Ramser, K.; Hanstorp, D. Optical manipulation for single-cell studies. J. Biophotonics 2010, 3, 187–206. [Google Scholar] [CrossRef] [PubMed]
- Ashkin, A. Acceleration and trapping of particles by radiation pressure. Phys. Rev. Lett. 1970, 24, 156. [Google Scholar] [CrossRef]
- Neuman, K.C.; Block, S.M. Optical trapping. Rev. Sci. Instrum. 2004, 75, 2787–2809. [Google Scholar] [CrossRef] [PubMed]
- Ashkin, A.; Dziedzic, J.M.; Bjorkholm, J.E.; Chu, S. Observation of a single-beam gradient force trap for dielectric particles. Opt. Lett. 1986, 11, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Neuman, K.C.; Liou, G.F.; Block, S.M.; Bergman, K. Characterization of photodamage induced by optical tweezers. In Conference on Lasers and Electro-Optics; Scifres, D., Weiner, A., Eds.; paper CTuR1; Optical Society of America: Washington, DC, USA, 1998. [Google Scholar]
- Maghelli, N.; Tolić-Nørrelykke, I.M. Optical trapping and laser ablation of microtubules in fission yeast. Methods Cell. Biol. 2010, 97, 173–183. [Google Scholar] [PubMed]
- Simmons, R.M.; Finer, J.T.; Chu, S.; Spudich, J.A. Quantitative measurements of force and displacement using an optical trap. Biophys. J. 1996, 70, 1813–1822. [Google Scholar] [CrossRef]
- Grimbergen, J.A.; Visscher, K.; de Gomes Mesquita, D.S.; Brakenhoff, G.J. Isolation of single yeast cells by optical trapping. Yeast 1993, 9, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Arai, F.; Ng, C.; Maruyama, H.; Ichikawa, A.; El-Shimy, H.; Fukuda, T. On chip single-cell separation and immobilization using optical tweezers and thermosensitive hydrogel. Lab Chip 2005, 5, 1399–1403. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.P.; Creely, C.M.; Volpe, G.; Grötsch, H.; Petrov, D. Real-time detection of hyperosmotic stress response in optically trapped single yeast cells using Raman microspectroscopy. Anal. Chem. 2005, 77, 2564–2568. [Google Scholar] [CrossRef] [PubMed]
- Aabo, T.; Banás, A.R.; Glückstad, J.; Siegumfeldt, H.; Arneborg, N. BioPhotonics workstation: A versatile setup for simultaneous optical manipulation, heat stress, and intracellular pH measurements of a live yeast cell. Rev. Sci. Instrum. 2011, 82, 083707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, E.; Scrimgeour, J.; Graneli, A.; Ramser, K.; Wellander, R.; Enger, J.; Hanstorp, D.; Goksör, M. Optical manipulation and microfluidics for studies of single cell dynamics. J. Opt. A Pure Appl. Opt. 2007, 9, S113–S121. [Google Scholar] [CrossRef]
- Dholakia, K.; Reece, P. Optical micromanipulation takes hold. Nanotoday 2006, 1, 18–27. [Google Scholar] [CrossRef]
- Castelein, M.; Rouxhet, P.G.; Pignon, F.; Magnin, A.; Piau, J.M. Single-cell adhesion probed in-situ using optical tweezers: A case study with Saccharomyces cerevisiae. J. Appl. Phys. 2012, 111, 114701. [Google Scholar] [CrossRef]
- Landenberger, B.; Höfemann, H.; Wadle, S.; Rohrbach, A. Microfluidic sorting of arbitrary cells with dynamic optical tweezers. Lab Chip 2012, 12, 3177–3183. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Merenda, F.; Piguet, J.; Salathé, R.P.; Vogel, H. Microfluidic array cytometer based on refractive optical tweezers for parallel trapping, imaging and sorting of individual cells. Lab Chip 2011, 11, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.M.; Castro, C.E.; Heath, R.J.; Cardenas, M.L.; Xavier, R.J.; Lang, M.J.; Vyas, J.M. Control and manipulation of pathogens with an optical trap for live cell imaging of intercellular interactions. PLoS ONE 2010, 5, e15215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, J.M.; Castro, C.E.; Heath, R.J.; Mansour, M.K.; Cardenas, M.L.; Xavier, R.J.; Lang, M.J.; Vyas, J.M. Use of an optical trap for study of host-pathogen interactions for dynamic live cell imaging. J. Vis. Exp. 2011, 53, 3123. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Sott, K.; Lundqvist, F.; Sveningsson, M.; Scrimgeour, J.; Hanstorp, D.; Goksör, M.; Granéli, A. A microfluidic device for reversible environmental changes around single cells using optical tweezers for cell selection and positioning. Lab Chip 2010, 10, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Arneborg, N.; Siegumfeldt, H.; Andersen, G.H.; Nissen, P.; Daria, V.R.; Rodrigo, P.J.; Glückstad, J. Interactive optical trapping shows that confinement is a determinant of growth in a mixed yeast culture. FEMS Microbiol. Lett. 2005, 245, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Charrunchon, S.; Limtrakul, J.; Chattham, N. Growth pattern of yeast cells studied under optical tweezers. In Frontiers in Optics; 2012/Laser Science XXVIII, OSA Technical Digest (Online); Paper FW1G.6; Optical Society of America: Rochester, NY, USA, 2012. [Google Scholar]
- Ando, J.; Bautista, G.; Smith, N.; Fujita, K.; Daria, V.R. Optical trapping and surgery of living yeast cells using a single laser. Rev. Sci. Instrum. 2008, 79, 103705. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sun, D. Automated transportation of single cells using robot-tweezer manipulation system. J. Lab. Autom. 2011, 16, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Huang, W.; Liu, Q.F.; Wu, Y.; Rao, Y.; Peng, G.D.; Lang, J.; Zhang, K. Graded-index optical fiber tweezers with long manipulation length. Opt. Express 2014, 22, 25267–25276. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, A.K.; van Niekerk, D.D.; Adiels, C.B.; du Preez, F.B.; Goksör, M.; Snoep, J.L. Sustained glycolytic oscillations in individual isolated yeast cells. FEBS J. 2012, 279, 2837–2847. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, A.K.; van Niekerk, D.D.; Adiels, C.B.; Kooi, B.; Goksör, M.; Snoep, J.L. Allosteric regulation of phosphofructokinase controls the emergence of glycolytic oscillations in isolated yeast cells. FEBS J. 2014, 281, 2784–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habaza, M.; Gilboa, B.; Roichman, Y.; Shaked, N.T. Tomographic phase microscopy with 180° rotation of live cells in suspension by holographic optical tweezers. Opt. Lett. 2015, 40, 1881–1884. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.; Wu, J.; Liu, G.W.; Keeler, E.G.; Pun, S.H.; Lin, L.Y. Photonic crystal optical tweezers with high efficiency for live biological samples and viability characterization. Sci. Rep. 2016, 6, 19924. [Google Scholar] [CrossRef] [PubMed]
- Tolić-Nørrelykke, I.M.; Munteanu, E.L.; Thon, G.; Oddershede, L.; Berg-Sørensen, K. Anomalous diffusion in living yeast cells. Phys. Rev. Lett. 2004, 93, 078102. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, L.; Tolić-Nørrelykke, I.M.; Stringari, C.; Antolini, R.; Pavone, F.S. Optical micromanipulations inside yeast cells. Appl. Opt. 2005, 44, 2001–2007. [Google Scholar] [CrossRef] [PubMed]
- Tolic-Nørrelykke, I.M.; Sacconi, L.; Stringari, C.; Raabe, I.; Pavone, F.S. Nuclear and division-plane positioning revealed by optical micromanipulation. Curr. Biol. 2005, 15, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Tejedor, V.; Burov, S.; Barkai, E.; Selhuber-Unkel, C.; Berg-Sørensen, K.; Oddershede, L.; Metzler, R. In vivo anomalous diffusion and weak ergodicity breaking of lipid granules. Phys. Rev. Lett. 2011, 106, 048103. [Google Scholar] [CrossRef] [PubMed]
- Mas, J.; Richardson, A.C.; Reihani, S.N.; Oddershede, L.B.; Berg-Sørensen, K. Quantitative determination of optical trapping strength and viscoelastic moduli inside living cells. Phys. Biol. 2013, 10, 046006. [Google Scholar] [CrossRef] [PubMed]
- Difato, F.; Pinato, G.; Cojoc, D. Cell signalling periments driven by optical manipulation. Int. J. Mol. Sci. 2013, 14, 8963–8984. [Google Scholar] [CrossRef] [PubMed]
- Kotsifaki, D.G.; Makropoulou, M.; Serafetinides, A. Near infrared optical tweezers and nanosecond ablation on yeast and algae cells. In Proceedings of the SPIE 17th International School on Quantum Electronics: Laser Physics and Applications, Nessebar, Bulgaria, 24–28 September 2012; Volume 8770.
- Oddershede, L.B. Force probing of individual molecules inside the living cell is now a reality. Nat. Chem. Biol. 2012, 8, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Norregaard, K.; Jauffred, L.; Berg-Sørensen, K.; Oddershede, L.B. Optical manipulation of single molecules in the living cell. Phys. Chem. Chem. Phys. 2014, 16, 12614–12624. [Google Scholar] [CrossRef] [PubMed]
- Toriello, N.M.; Douglas, E.S.; Mathies, R.A. Microfluidic device for electric field-driven single-cell capture and activation. Anal. Chem. 2005, 77, 6935–6941. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Tsubouchi, T.; Usui, K.; Uematsu, K.; Tame, A.; Nogi, Y.; Ohta, Y.; Hatada, Y.; Kato, C.; Miwa, T.; et al. Involvement of flocculin in negative potential-applied ITO electrode adhesion of yeast cells. FEMS Yeast Res. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Huang, H.; Chen, L.; Li, X.; Ge, Z.; Chen, T.; Yang, Z.; Sun, L. Dielectrophoresis for bioparticle manipulation. Int. J. Mol. Sci. 2014, 15, 18281–18309. [Google Scholar] [CrossRef] [PubMed]
- Norde, W. Colloids and Interfaces in Life Sciences; Marcel Dekker: Monticello, NY, USA, 2003. [Google Scholar]
- Jubery, T.Z.; Srivastava, S.K.; Dutta, P. Dielectrophoretic separation of bioparticles in microdevices: A review. Electrophoresis 2014, 35, 691–713. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.H.; Xuan, X.C.; Kang, Y.J.; Li, D.Q. Effects of dc-dielectrophoretic force on particle trajectories in microchannels. J. Appl. Phys. 2006, 99, 064702. [Google Scholar] [CrossRef]
- Khoshmanesh, K.; Nahavandi, S.; Baratchi, S.; Mitchell, A.; Kalantar-zadeh, K. Dielectrophoretic platforms for bio-microfluidic systems. Biosens. Bioelectron. 2011, 26, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Koklu, M.; Park, S.; Pillai, S.D.; Beskok, A. Negative dielectrophoretic capture of bacterial spores in food matrices. Biomicrofluidics 2010, 4. [Google Scholar] [CrossRef] [PubMed]
- Cheng, I.F.; Froude, V.E.; Zhu, Y.; Chang, H.C.; Chang, H.C. A continuous high-throughput bioparticle sorter based on 3D traveling-wave dielectrophoresis. Lab Chip 2009, 9, 3193–3201. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.P.; Westervelt, R.M. Dielectrophoresis tweezers for single cell manipulation. Biomed. Microdevices 2006, 8, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Cetin, B.; Kang, Y.; Wu, Z.; Li, D. Continuous particle separation by size via AC-dielectrophoresis using a lab-on-a-chip device with 3-D electrodes. Electrophoresis 2009, 30, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Docoslis, A.; Kalogerakis, N.; Behie, L.A. Dielectrophoretic forces can be safely used to retain viable cells in perfusion cultures of animal cells. Cytotechnology 1999, 30, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Pethig, R.; Menachery, A.; Pells, S.; de Sousa, P. Dielectrophoresis: A review of applications for stem cell research. J. Biomed. Biotechnol. 2010, 2010, 182581. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, M.; Bougot-Robin, K.; Cao, W.; Chau, I.Y.Y.; Li, W.; Wen, W. High-throughput particle manipulation by hydrodynamic, electrokinetic, and dielectrophoretic effects in an integrated microfluidic chip. Biomicrofluidics 2013, 7, 24106. [Google Scholar] [CrossRef] [PubMed]
- Li, P.C.; Harrison, D.J. Transport, manipulation, and reaction of biological cells on-chip using electrokinetic effects. Anal. Chem. 1997, 69, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Osaki, T.; Kawano, R.; Kamiya, K.; Miki, N.; Takeuchi, S. Round-tip dielectrophoresis-based tweezers for single micro-object manipulation. Biosens. Bioelectron. 2013, 47, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.; Voldman, J. Dielectrophoretic traps for single-particle patterning. Biophys. J. 2005, 88, 2193–2205. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hunt, T.P.; Westervelt, R.M. Magnetic and electric manipulation of a single cell in fluid. Mater. Res. Symp. Proc. 2004, 820. [Google Scholar] [CrossRef]
- Lee, H.; Purdon, A.M.; Westervelt, R.M. Micromanipulation of biological systems with microelectromagnets. IEEE Trans. Magn. 2004, 40, 2991–2993. [Google Scholar] [CrossRef]
- Jang, L.S.; Huang, P.H.; Lan, K.C. Single-cell trapping utilizing negative dielectrophoretic quadrupole and microwell electrodes. Biosens. Bioelectron. 2009, 24, 3637–3644. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.M. Positioning and levitation media for the separation of biological cells. Ind. Appl. IEEE Trans. 2001, 37, 1468–1475. [Google Scholar] [CrossRef]
- Gagnon, Z.; Mazur, J.; Chang, H.C. Glutaraldehyde enhanced dielectrophoretic yeast cell separation. Biomicrofluidics 2009, 3, 44108. [Google Scholar] [CrossRef] [PubMed]
- Fatoyinbo, H.O.; Kamchis, D.; Whattingham, R.; Ogin, S.L.; Hughes, M.P. A high-throughput 3-D composite dielectrophoretic separator. IEEE Trans. Biomed. Eng. 2005, 52, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Urdaneta, M.; Smela, E. Multiple frequency dielectrophoresis. Electrophoresis 2007, 28, 3145–3155. [Google Scholar] [CrossRef] [PubMed]
- Salomon, S.; Leichlé, T.; Nicu, L. A dielectrophoretic continuous flow sorter using integrated microelectrodes coupled to a channel constriction. Electrophoresis 2011, 32, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hölzel, R.; Pethig, R.; Wang, X.B. Differences in the AC electrodynamics of viable and non-viable yeast cells determined through combined dielectrophoresis and electrorotation studies. Phys. Med. Biol. 1992, 37, 1499–1517. [Google Scholar] [CrossRef] [PubMed]
- Hölzel, R. Electrorotation of single yeast cells at frequencies between 100 Hz and 1.6 GHz. Biophys. J. 1997, 73, 1103–1109. [Google Scholar] [CrossRef]
- Fatoyinbo, H.O.; Hoettges, K.F.; Hughes, M.P. Rapid-on-chip determination of dielectric properties of biological cells using imaging techniques in a dielectrophoresis dot microsystem. Electrophoresis 2008, 29, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soffe, R.; Tang, S.Y.; Baratchi, S.; Nahavandi, S.; Nasabi, M.; Cooper, J.M.; Mitchell, A.; Khoshmanesh, K. Controlled rotation and vibration of patterned cell clusters using dielectrophoresis. Anal. Chem. 2015, 87, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.P.; Issadore, D.; Westervelt, R.M. Integrated circuit/microfluidic chip toprogrammably trap and move cells and droplets with dielectrophoresis. Lab. Chip 2008, 8, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Witek, M.A.; Wei, S.; Vaidya, B.; Adams, A.A.; Zhu, L.; Stryjewski, W.; McCarley, R.L.; Soper, S.A. Cell transport via electromigration in polymer-based microfluidic devices. Lab Chip 2004, 4, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Frenea-Robin, M.; Chetouani, H.; Haddour, N.; Rostaing, H.; Laforet, J.; Reyne, G. Contactless diamagnetic trapping of living cells onto a micromagnet array. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2008, 2008, 3360–3363. [Google Scholar] [PubMed]
- Yasukawa, T.; Nagamine, K.; Horiguchi, Y.; Shiku, H.; Koide, M.; Itayama, T.; Shiraishi, F.; Matsue, T. Electrophoretic cell manipulation and electrochemical gene-function analysis based on a yeast two-hybrid system in a microfluidic device. Anal. Chem. 2008, 80, 3722–3727. [Google Scholar] [CrossRef] [PubMed]
- Moncada-Hernandez, H.; Baylon-Cardiel, J.L.; Pérez-González, V.H.; Lapizco-Encinas, B.H. Insulator-based dielectrophoresis of microorganisms: Theoretical and experimental results. Electrophoresis 2011, 32, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.; Nakajima, M.; Tajima, H.; Fukuda, T. Fabrication of microstructures embedding controllable particles inside dielectrophoretic microfluidic devices. Int. J. Adv. Robot. Syst. 2013, 10, 132. [Google Scholar] [CrossRef]
- Shi, X.; Shi, Z.; Wang, D.; Ullah, M.W.; Yang, G. Microbial cells with a Fe3O4 doped hydrogel extracellular matrix: Manipulation of living cells by magnetic stimulus. Macromol. Biosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Winkleman, A.; Gudiksen, K.L.; Ryan, D.; Whitesides, G.M.; Greenfield, D.; Prentiss, M. A magnetic trap for living cells suspended in a paramagnetic buffer. Appl. Phys. Lett. 2004, 85, 2411. [Google Scholar] [CrossRef]
- Neuman, K.C.; Nagy, A. Single-molecule force spectroscopy: Optical tweezers, magnetic tweezers and atomic force microscopy. Nat. Methods 2008, 5, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.K.; Cribb, J.; Desai, K.V.; Vicci, L.; Wilde, B.; Keller, K.; Taylor, R.M.; Haase, J.; Bloom, K.; O’Brien, E.T.; et al. Thin-foil magnetic force system for high-numerical-aperture microscopy. Rev. Sci. Instrum. 2006, 77. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Skoko, D.; Marko, J.F. Near-field-magnetic-tweezer manipulation of single DNA molecules. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2004, 70, 011905. [Google Scholar] [CrossRef] [PubMed]
- Chacko, J.V.; Zanacchi, F.C.; Diaspro, A. Probing cytoskeletal structures by coupling optical superresolution and AFM techniques for a correlative approach. Cytoskeleton (Hoboken) 2013, 70, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Chacko, J.V.; Harke, B.; Canale, C.; Diaspro, A. Cellular level nanomanipulation using atomic force microscope aided with superresolution imaging. J. Biomed. Opt. 2014, 19, 105003. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, H.; Hiraoka, Y.; Haraguchi, T. A method of correlative light and electron microscopy for yeast cells. Micron 2014, 61, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Iwamoto, M.; Haraguchi, T. Live correlative light-electron microscopy to observe molecular dynamics in high resolution. Microscopy (Oxf.) 2016, 65, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.; Hagen, C.; Grünewald, K.; Kaufmann, R. Towards correlative super-resolution fluorescence and electron cryo-microscopy. Biol. Cell 2016, 108, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, J.J.; Lipke, P.N.; Beaussart, A.; El Kirat Chatel, S.; Dupres, V.; Alsteens, D.; Dufrêne, Y.F. Atomic force microscopy—Looking at mechanosensors on the cell surface. J. Cell Sci. 2012, 125, 4189–4195. [Google Scholar] [CrossRef] [PubMed]
- Conroy, R. Force spectroscopy with optical and magnetic tweezers. In Handbook of Molecular Force Spectroscopy; Noy, A., Ed.; Springer: New York, NY, USA, 2008; pp. 23–96. [Google Scholar]
- Alonso-Sarduy, L.; de Los Rios, P.; Benedetti, F.; Vobornik, D.; Dietler, G.; Kasas, S.; Longo, G. Real-time monitoring of protein conformational changes using a nano-mechanical sensor. PLoS ONE 2014, 9, e103674. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yeast Type | AFM Analysis | Objective | Refs |
|---|---|---|---|
| C. albicans | Imaging, cell surface elasticity | Effect of antifungal caspofungin | [16] |
| Imaging, cell elasticity | Imaging mode evaluation | [17] | |
| Imaging, force spectroscopy using concanavalin A-functionalised tips | Mapping of adhesive properties | [18] | |
| Candida parapsilosis | Imaging, adhesion force | Surface morphological characterisation | [19] |
| S. cerevisiae | Imaging | Immobilisation method | [8] |
| Imaging | Immobilisation method | [9] | |
| Imaging, force spectroscopy using concanavalin A-functionalised tips | Mapping cell wall polysaccharides | [20] | |
| Imaging, cell elasticity | Mapping of cell elasticity | [21] | |
| Imaging | Cell surface change on thermal and osmotic stress | [22] | |
| Imaging, motion analysis | Nanomechanical motion analysis | [23] | |
| Imaging | Effect of electromagnetic field and antifungal nystatin on the cell wall | [24] | |
| Imaging, cell elasticity | Immobilisation method | [10] | |
| Imaging | Immobilisation method | [11] | |
| Imaging, cell surface elasticity | Effect of antifungal caspofungin | [16] | |
| Sc. pombe | Imaging | Cell surface change on thermal and osmotic stress | [22] |
| Technique | Description | Spatial Resolution | Timescale |
|---|---|---|---|
| Fluorescence imaging with one-nanometer accuracy (FIONA) | Localises and tracks single-molecule emitters by finding the centre of their diffraction-limited point-spread function (PSF). | ~1.5 nm | ~0.3 ms |
| Single-molecule high-resolution colocalisation (SHREC) | Two-colour version of FIONA. Two fluorescent probes with different spectra are imaged separately and then localised and mapped onto the plane of the microscope. | <10 nm | ~1 s per frame |
| Single-molecule high-resolution imaging with photobleaching (SHRImP) | Uses the strategy wherein, upon photobleaching of two or more closely-spaced identical fluorophores, their position is sequentially determined by FIONA, starting from the last bleached fluorophore. | ~5 nm | ~0.5 s per frame |
| Nanometer-localised multiple single-molecules (NALMS) | Uses a similar principle to single-molecule high-resolution imaging with photobleaching to measure distances between identical fluorescent probes that overlap within a diffraction-limited spot. | ~8 nm | ~1 s per frame |
| Photoactivatable localization microscopy (PALM) | Serially photoactivates and photodeactivates many sparse subsets of photoactivatable fluorophores to produce a sequence of images that are combined into a super-resolution composite. | ~2 nm | ~1 min |
| PALM with independently running acquisition (PALMIRA) | Records non-triggered spontaneous off–on–off cycles of photoswitchable fluorophores without synchronising the detector to reach faster acquisition. | ~50 nm | ~2.5 min |
| Single particle tracking PALM | Combines PALM with live-cell single fluorescent particle tracking. | ||
| Stimulated emission depletion (STED) | Reduces the excitation volume below that dictated by the diffraction limit by coaligning one beam of light capable of fluorophore excitation with another that induces de-excitation by stimulated emission. | ~16 nm | ~10 min |
| Stochastic optical reconstruction microscopy (STORM) | Small sub-populations of photoswitchable fluorophores are turned on and off using light of different colours, permitting the localisation of single molecules. Repeated activation cycles produce a composite image of the entire sample. | <20 nm | ~mins |
| Cell Type | Interacting Molecule 1 | Interacting Molecule 2 | Rupture Force (pN) | Refs |
|---|---|---|---|---|
| C. albicans | Als5p | Fibronectin | 2800 ± 600 | [103] |
| S. cerevisiae | Prion protein Sup35 hexapeptide | Prion protein Sup35 hexapeptide | — | [104] |
| Prion protein Sup35 hexapeptide antiparallel hairpin structure | Prion protein Sup35 hexapeptide antiparallel hairpin structure | 32–134 | [105] | |
| Nucleoporin | Nucleoporin | — | [106] | |
| Nucleoporin | Importin | — | ||
| Flo1p | Flo1p | 300 (100–600) | [107] |
| Cell Type | Cell Receptor | Ligand | Rupture Force (pN) | Refs |
|---|---|---|---|---|
| C. albicans | Cell surface β-mannan | Anti-β-1,2-mannoside antibodies | 41 ± 14 | [108] |
| Cell surface β-glucans | Anti-β-1,3-glucan antibodies | 38 ± 10 | [108] | |
| Cell wall chitin | WGA 1 lectin | 65 ± 19 | [108] | |
| Cell wall | Streptococcus mutans exoenzyme glycosyltransferase B | 1000–2000 | [109] | |
| C. glabrata | Epa6p | Hydrophobic surface | - | [110] |
| Cell surface β-mannan | Anti-β-1,2-mannoside antibodies | 54 ± 9 | [108] | |
| Cell surface β-glucans | Anti-β-1,3-glucan antibodies | 41 ± 8 | [108] | |
| Cell wall chitin | WGA 1 lectin | 41 ± 8 | [108] | |
| S. cerevisiae | Cell surface α-mannan | Con A 2 lectin | 75-200 | [111] |
| Cell surface α-mannan | Con A 2 lectin | 92 ± 35 | [108] | |
| Cell surface β-glucans | Anti-β-1,3-glucan antibodies | 42 ± 7 | [108] | |
| Cell wall chitin | WGA 1 lectin | 54 ± 19 | [108] | |
| Wsc1p-His-tagged | NTA-Ni2+ | - | [112,113,114] | |
| HA 3-tagged Ccw12p | Anti-HA antibody | 69.3 ± 31.4 | [115] | |
| Ste2p | α-factor | 250 | [116] | |
| S. pastorianus | Flo protein | Glucose | 121 ± 53 | [111] |
| Flo protein | Con A 2 | 117 ± 41 | [111] |
| Cell Type | Interaction Partner | Variables | Refs |
|---|---|---|---|
| C. albicans | Staphylococcus aureus | Deletion of ALS3 (adhesion gene) | [123] |
| Hydrophobic DDP 1 coated surface | Surface hydrophilicity, hydrophobicity, deletion of HGC1, compared to S. cerevisiae | [124] | |
| C. albicans hyphae | Deletion of ALS3 and ALS1 (adhesion genes) | [125] | |
| DC-SIGN 2 | Differences in the N-mannan structure of the cell wall | [118] | |
| C. glabrata | Adhesin Epa6p | Surface hydrophilicity, hydrophobicity, expressed and deleted EPA6 | [110] |
| S. cerevisiae | Abiotic surface | Surface hydrophilicity, hydrophobicity BSA coating, life cycle stage, glutaraldehyde-treated cells | [126] |
| Silica surface | Different silica with defined roughness | [127] | |
| Methacrylate polymers surface | Polymer imprinted and non-imprinted surface | [128] | |
| Bare and polydopamine-coated glass | Polydopamine coating | [129] |
| Yeast Type | Cell Patterning Method | Issue Addressed | Refs |
|---|---|---|---|
| S. cerevisiae | Patterning on adhesive micropatterns | Microcontact printing of concanavalin A | [157] |
| Mechanical cell patterning in microfluidic microchambers | Monitoring dynamics of single-cell gene expression | [158] | |
| Robotic cell printing | Systematic profiling of cellular phenotypes | [159] | |
| Mechanical cell patterning using trap barriers | Single cell gene expression analysis | [160] | |
| Robotic cell printing | Localisation of the yeast proteome during polarised growth | [161] | |
| Mechanical cell patterning in microfluidic microchambers | Quantitative analysis of the yeast pheromone signalling response | [162] | |
| Patterning on adhesive micropatterns | Microcontact printing of biotinylated bovine serum albumin | [163] | |
| Mechanical cell patterning using trap barriers | Whole lifespan microscopic observation | [164] | |
| Mechanical cell patterning in microfluidic single-cell microwells | Real-time cellular responses of the mating MAPK pathway | [165] | |
| Mechanical and chemical patterning in microchambers | Molecular phenotyping of aging in single cells | [166] | |
| Mechanical cell patterning using trap barriers | Single cell analysis of yeast replicative aging | [167] | |
| Mechanical cell patterning in elongated cavities | Monitoring the dynamics of cell division | [168] | |
| Mechanical cell patterning using trap barriers | Studying ageing and dynamic single-cell responses | [169] | |
| Patterning in microcavity array by negative pressure, and embedded in agarose gel layer | Long-term single cell growth observation | [170] | |
| Mechanical cell patterning using trap barriers | Automated measurements of single-cell aging | [171] | |
| Mechanical cell patterning using trap barriers | High-throughput analysis of yeast replicative aging | [172] | |
| Sc. pombe | Mechanical patterning in culture microchambers | Mechanical mechanisms redirecting cell polarity and cell shape in fission yeast | [173] |
| Mechanical patterning in single-cell microwells | Determination of the mechanical forces involved in cell growth | [174] | |
| Mechanical patterning in microchambers | Time-lapse fluorescence observation of the effect of a microtubule-inhibiting drug | [175] | |
| Mechanical trapping in single-cell cavities | Fission yeast synchronisation | [176] | |
| Mechanical barrier single-cell trapping | Lon-term observation using super-resolution fluorescence microscopy | [177] | |
| Mechanical patterning in chemostat microchambers | Long-term single-cell analysis | [178] | |
| Mechanical patterning in culture microchambers | Studies of cellular aging | [179] |
| Yeast Type | Issue Addressed | Refs |
|---|---|---|
| C. albicans | Control and manipulation of pathogenic yeast for live cell imaging and interaction with host cells | [237,238] |
| Hanseniaspora uvarum and S. cerevisiae | Confinement of an individual H. uvarum cell by S. cerevisiae cells increases the average generation time | [240] |
| S. bayanus | Study of growth pattern of cells under line optical tweezers generated by time-shared multiple optical traps | [241] |
| S. cerevisiae | On-chip single-cell separation and immobilisation using optical manipulation and thermosensitive hydrogel | [229] |
| Real-time detection of hyperosmotic stress response in optically trapped single yeast cells using Raman microspectroscopy | [230] | |
| Optical manipulation of cells to microscopically observe environmentally-induced size modulations and spatial localisation of GFP-tagged proteins to elucidate various signalling pathways | [232] | |
| Optical trapping and surgery of living cells using two operational modes of a single laser | [242] | |
| Selection and positioning of single cells combined with microscopy analysis in a microfluidic channel; cycling of GFP-tagged Mig1p and Msn1p between the cytosol and nucleus | [239] | |
| Optical trapping and fluorescence microscopy investigation of the internal pH response and membrane integrity with increasing temperature | [232] | |
| Automated transportation of single cells | [243] | |
| Development of a microfluidic array cytometer based on refractive optical tweezers for parallel trapping, imaging, and sorting of individual cells | [236] | |
| Microfluidic sorting of arbitrary cells with dynamic optical tweezers | [235] | |
| Development of graded-index optical fibre tweezers with long manipulation length | [244] | |
| Position yeast cells in a microfluidic chamber to study glycolytic oscillations | [245,246] | |
| Tomographic phase microscopy with live cell rotation using holographic optical tweezers | [247] | |
| Development of a photonic crystal optical tweezer to trap an array of yeast cells | [248] | |
| Sc. pombe | Displacement of the lipid granules | [249] |
| Displacement of the nucleus | [250,251] | |
| Laser ablation of microtubules in vivo | [226] | |
| In vivo anomalous diffusion and weak ergodicity breaking of lipid granules | [252] | |
| Quantitative determination of optical trapping strength and viscoelastic moduli inside living cells | [253] |
| Yeast Type | Manipulation Method | Issue Addressed | Refs |
|---|---|---|---|
| S. cerevisiae | Electrophoresis and electroosmosis | Cell transport in microfluidic channels | [272] |
| Dielectrophoresis | Live and dead cell separation | [278] | |
| Magnetic patterning | Demonstration of magnetic micromanipulation of magnetically labelled cells | [276] | |
| Electroosmosis | Cell transport via electromigration in polymer-based microfluidic devices | [288] | |
| Dielectrophoresis (AC DEP) | Sorting live and dead cells | [280] | |
| Dielectrophoresis (AC DEP) | DEP tweezer for single cell manipulation | [267] | |
| Dielectrophoresis | Multiple frequency DEP separation and trapping of live and dead cells | [281] | |
| Diamagnetic trapping | Cell magnetic trapping in an array using a CoPt micromagnet array | [289] | |
| Magnetophoresis | Contactless diamagnetic trapping of cells onto a micromaget array | [289] | |
| Electrophoresis | Electrophoretic cell manipulation in a microfluidic device | [290] | |
| Dielectrophoresis (AC DEP) | Separation of yeast cells from blood cells in a microfluidic chip | [268] | |
| Dielectrophoresis | Live and dead cell separation | [279] | |
| Dielectrophoresis | Microfluidic chip for guiding cells by AC electrothermal effect and capturing by nDEP trap | [277] | |
| Dielectrophoresis (DC DEP) | Separation of a mixture of S. cerevisiae and Escherichia coli cells | [291] | |
| Dielectrophoresis (AC/DC DEP) | Sorting live and dead cells | [282] | |
| Dielectrophoresis, electroosmosis, electrophoresis | High-throughput trapping of cells, separation of live and dead cells | [271] | |
| Dielectrophoresis | Cell manipulation and immobilisation using photo-crosslinkable resin inside microfluidic devices | [292] | |
| Dielectrophoresis | Controlled rotation and vibration of cell clusters | [286] | |
| Magnetic manipulation | Magnetic manipulation of Fe3O4-doped hydrogel-coated cells | [293] |
| Characteristic | AFM | Electron Microscopy (SEM, TEM) | Super-Resolution Fluorescence Microscopy (PALM, STORM, SIM) |
|---|---|---|---|
| Resolution | ~10 nm 1 | ~1–10 nm | ~5–50 nm |
| Live cell | Yes | No | Yes |
| Sample preparation requirement | Little | Little to substantial | Little to moderate |
| Sample preparation time | 10 min–1 d | 2 h–5 d | 30 min |
| Image acquisition time | ~5 min | 5–10 min | Up to 24 h |
| Equipment cost | €150,000–350,000 | €500,000 | €250,000–500,000 |
| Operational costs | Low | High | Moderate |
| Advantages | Localisation (and force spectroscopy) of single proteins; observation of dynamic processes; various environments (temperature, liquid, air, etc.) | Imaging of the cell ultrastructure at very high resolution | Time resolution. |
| Disadvantages | Only the cell surface is analysed; only one single cell at a time; slow temporal resolution; various sources of artifacts, such as cell or tip alteration | Fixation artifacts; no dynamics; no information on physical properties of proteins | Labelling is required |
| Characteristic | Optical Tweezers | Magnetic Tweezers | AFM | Micropipette |
|---|---|---|---|---|
| Type | Point | Global/point | Point | Point |
| Non-contact | Non-contact | Contact | Contact | |
| Spatial resolution (nm) | 0.1–2 | 5–10 | 0.5–1 | - |
| Temporal resolution (s) | 10−4 | 10−1–10−2 | 10−3 | - |
| Stiffness (pN nm−1) | 0.005–1 | 10−3–10−6 | 10–105 | 0.01–1000 |
| Force range (pN) | 0.1–100 | 10−3–102 | 10–104 | 1–1000 |
| Probe size (µm) | 0.25–5 | 0.5–5 | 100–250 | |
| Energy dissipation | Yes | No | No | No |
| Surface considerations | No | No | Yes | Yes |
| Features | Low noise and drift dumbbell geometry; access inside a cell | Force clamp, bead rotation, specific interactions; access inside a cell | High-resolution imaging | Controlled deposition/transfer of selected cells |
| Limitations | Photodamage, sample heating, non specific | No manipulation (force hysteresis) | Large high-stiffness probe, large minimal force, non specific | Low throughput |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willaert, R.; Kasas, S.; Devreese, B.; Dietler, G. Yeast Nanobiotechnology. Fermentation 2016, 2, 18. https://doi.org/10.3390/fermentation2040018
Willaert R, Kasas S, Devreese B, Dietler G. Yeast Nanobiotechnology. Fermentation. 2016; 2(4):18. https://doi.org/10.3390/fermentation2040018
Chicago/Turabian StyleWillaert, Ronnie, Sandor Kasas, Bart Devreese, and Giovanni Dietler. 2016. "Yeast Nanobiotechnology" Fermentation 2, no. 4: 18. https://doi.org/10.3390/fermentation2040018
APA StyleWillaert, R., Kasas, S., Devreese, B., & Dietler, G. (2016). Yeast Nanobiotechnology. Fermentation, 2(4), 18. https://doi.org/10.3390/fermentation2040018