
Mixed-Culture Metagenomics of the Microbes Making Sour Beer



Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction and Sequencing
2.3. Bioinformatics Analyses
3. Results
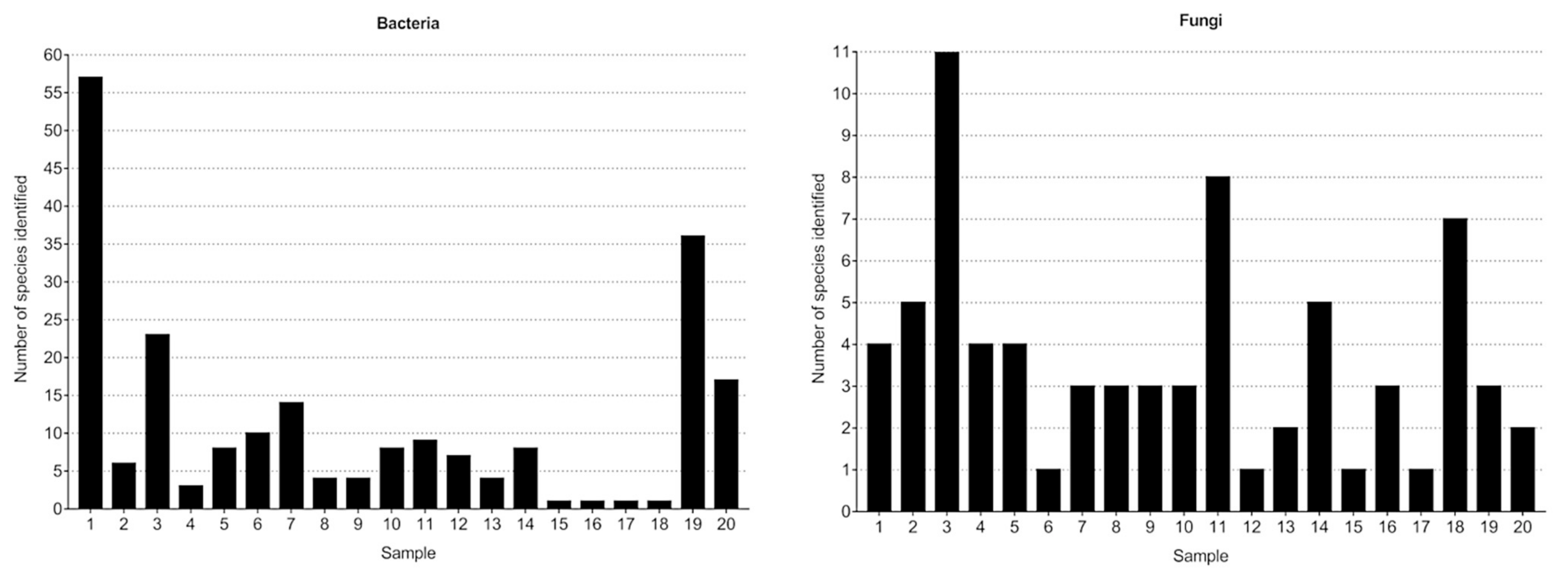
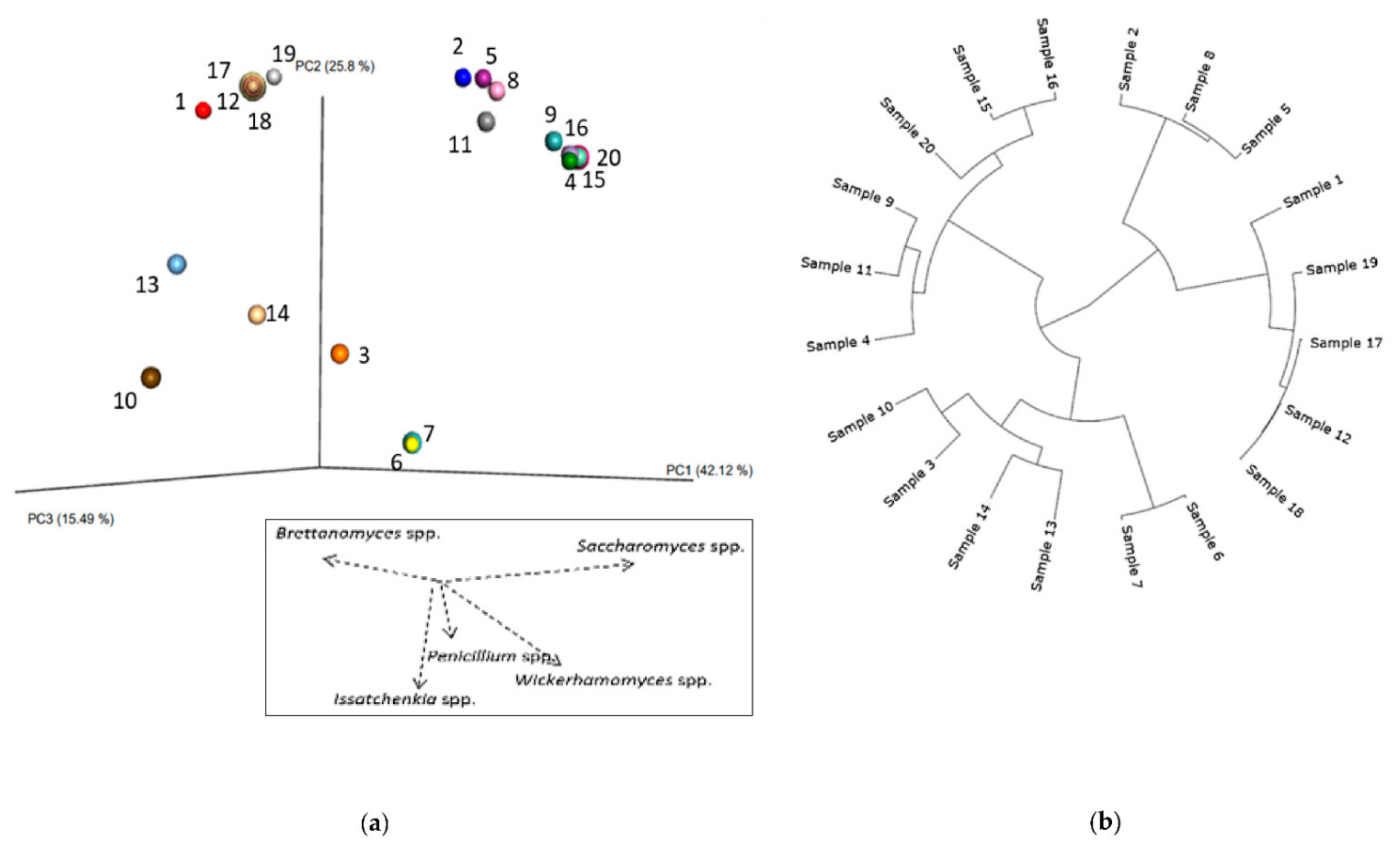
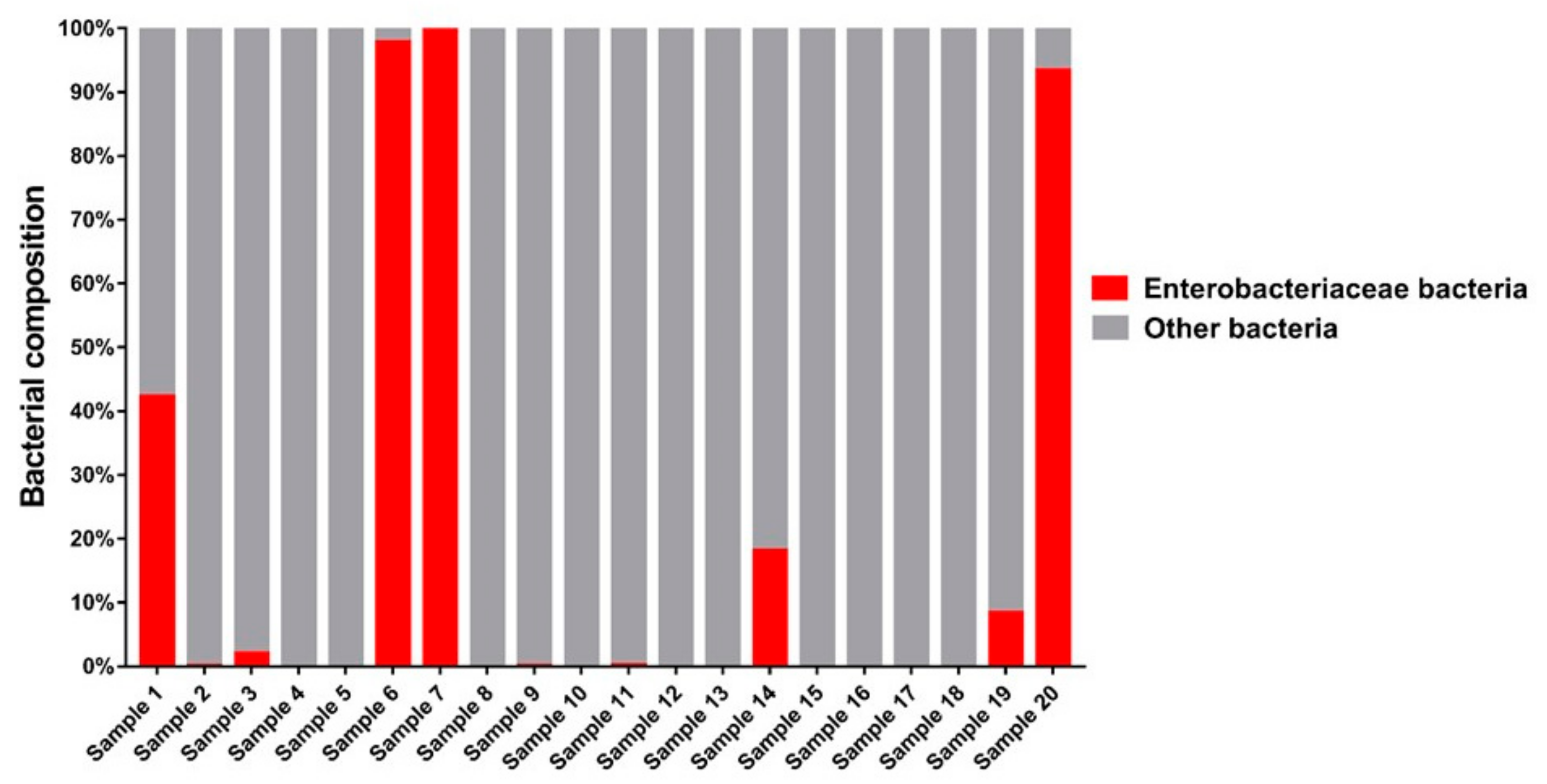
3.1. Bacteria—16S rRNA V3-V4 Region Analysis
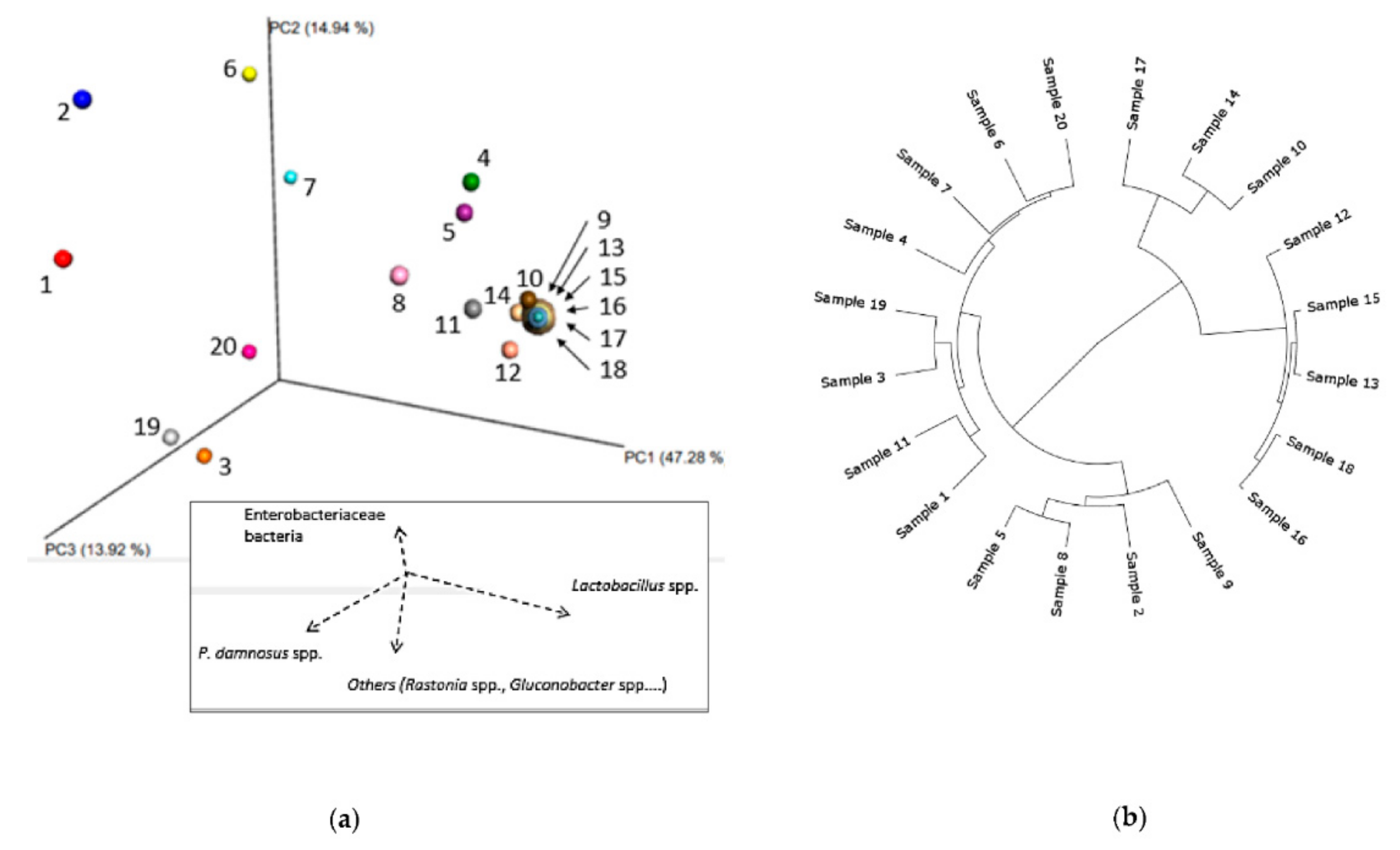
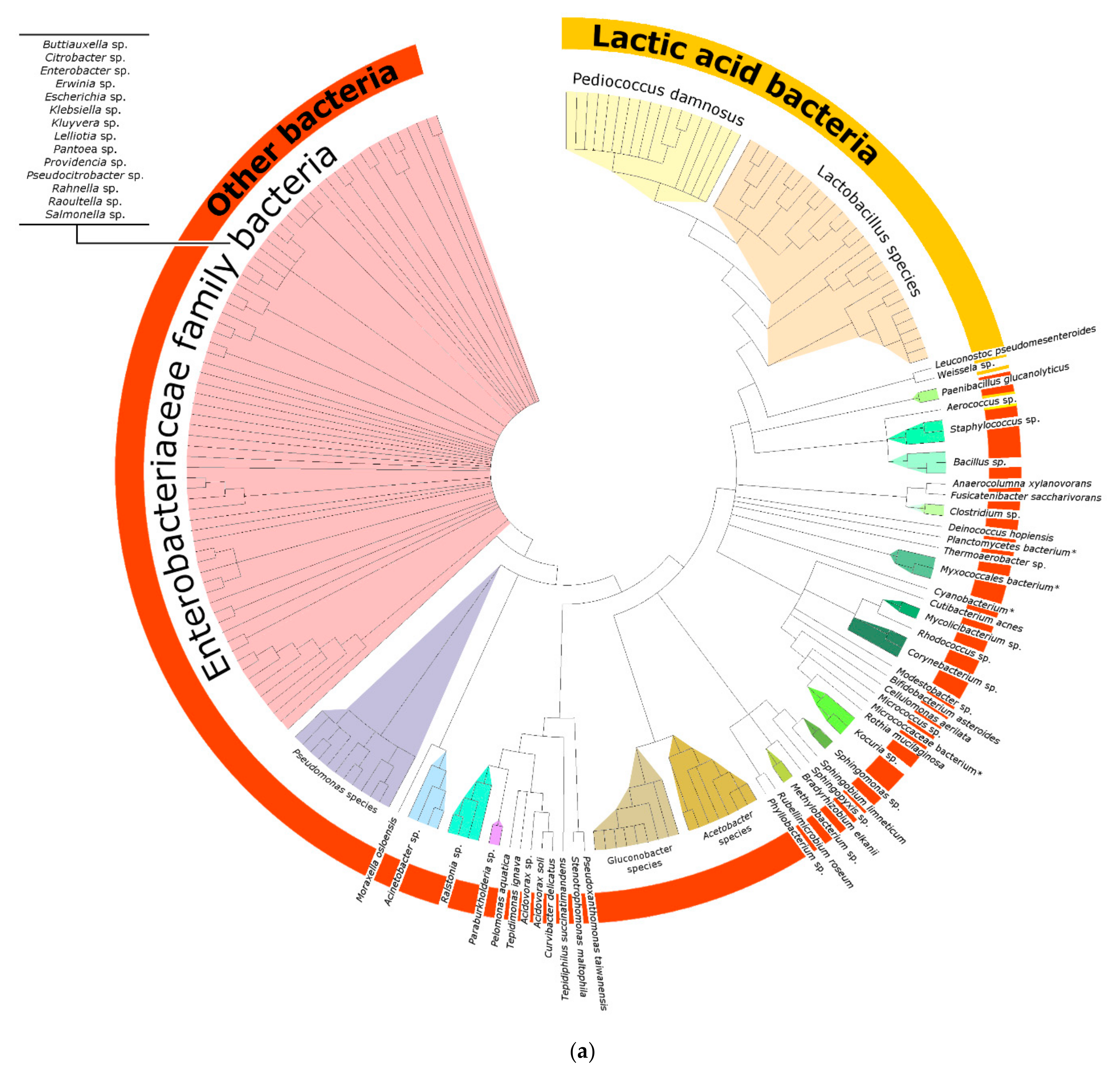
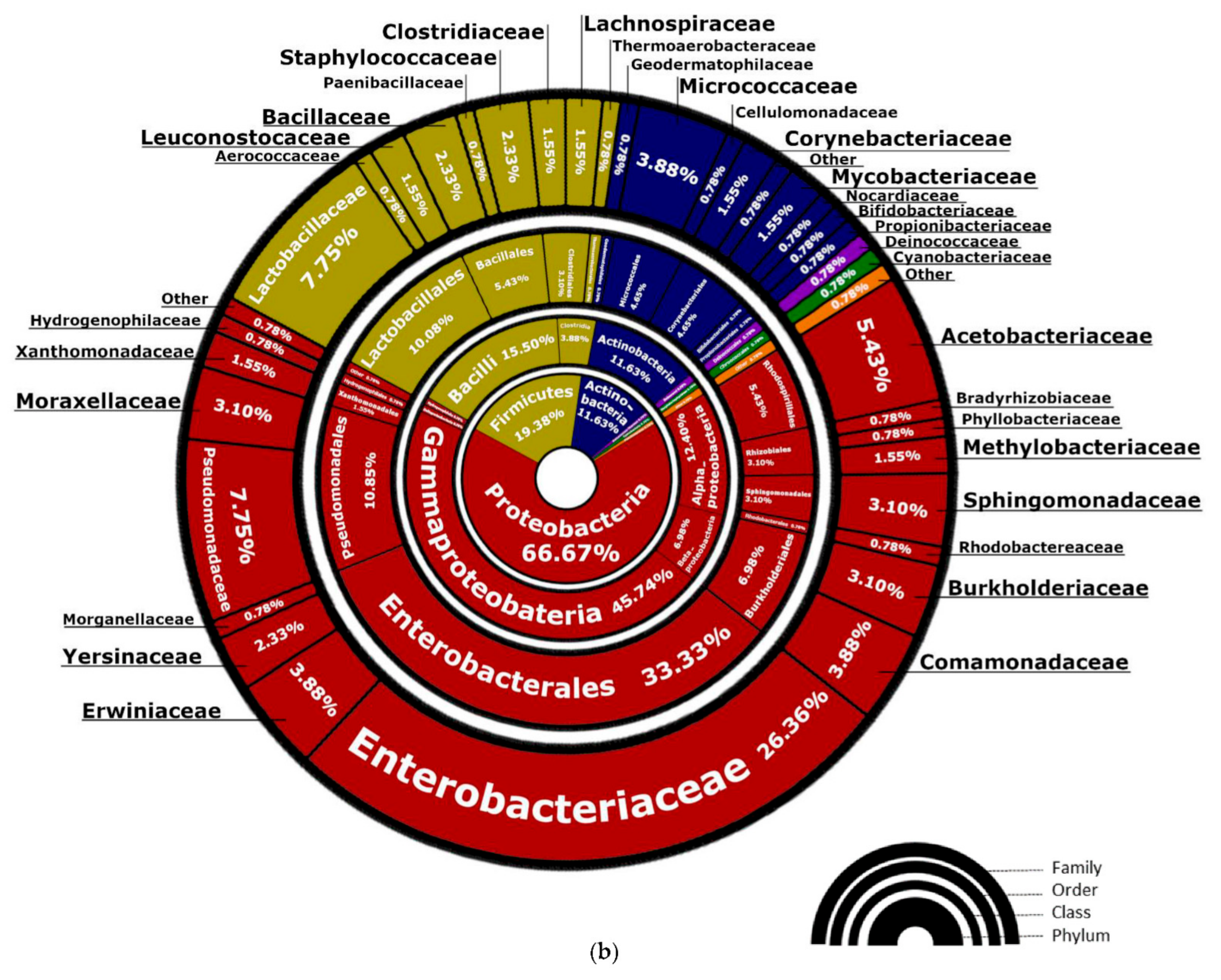
3.1.1. Bacterial Composition
3.1.2. Phylogenetic Analysis
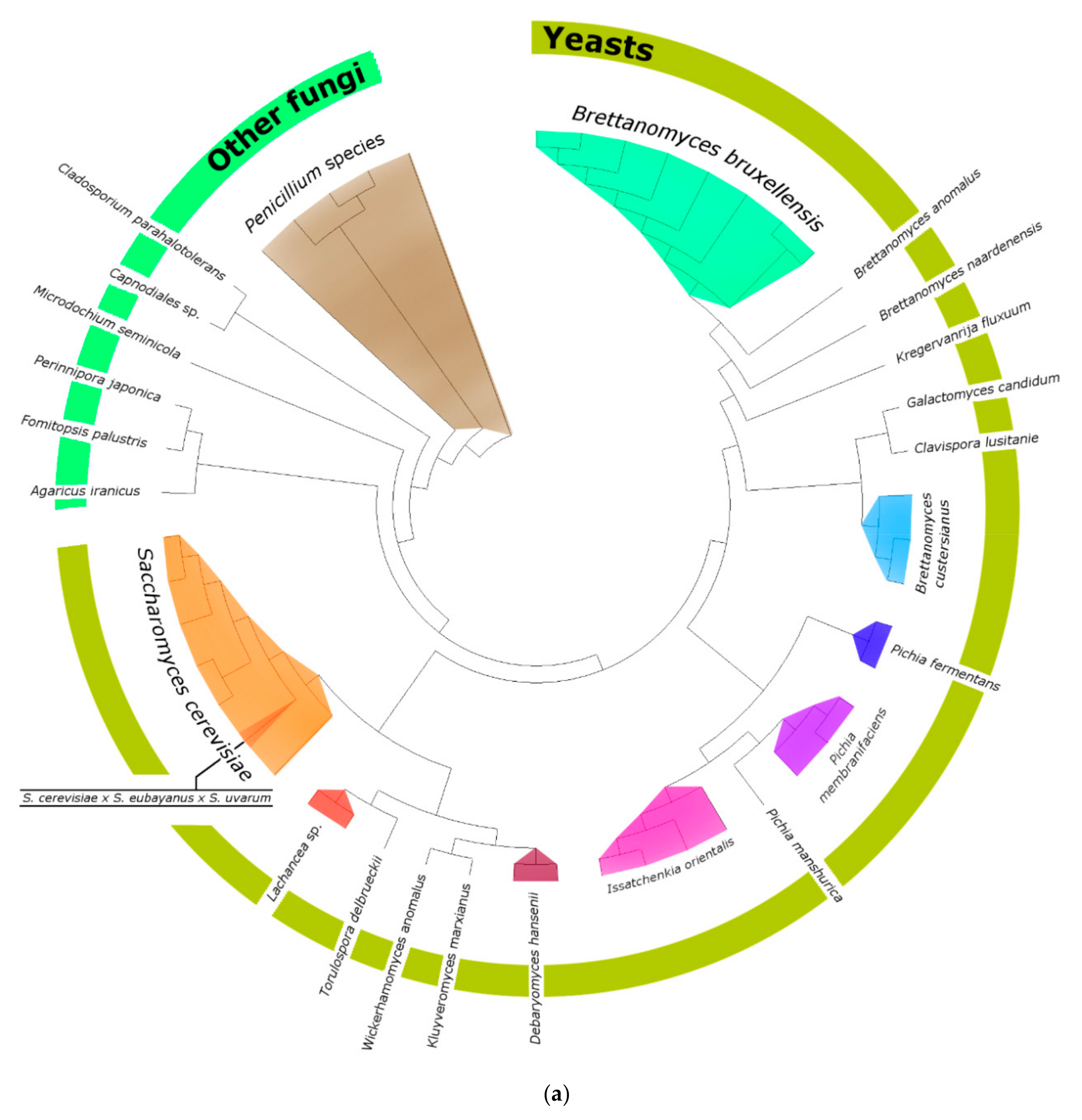
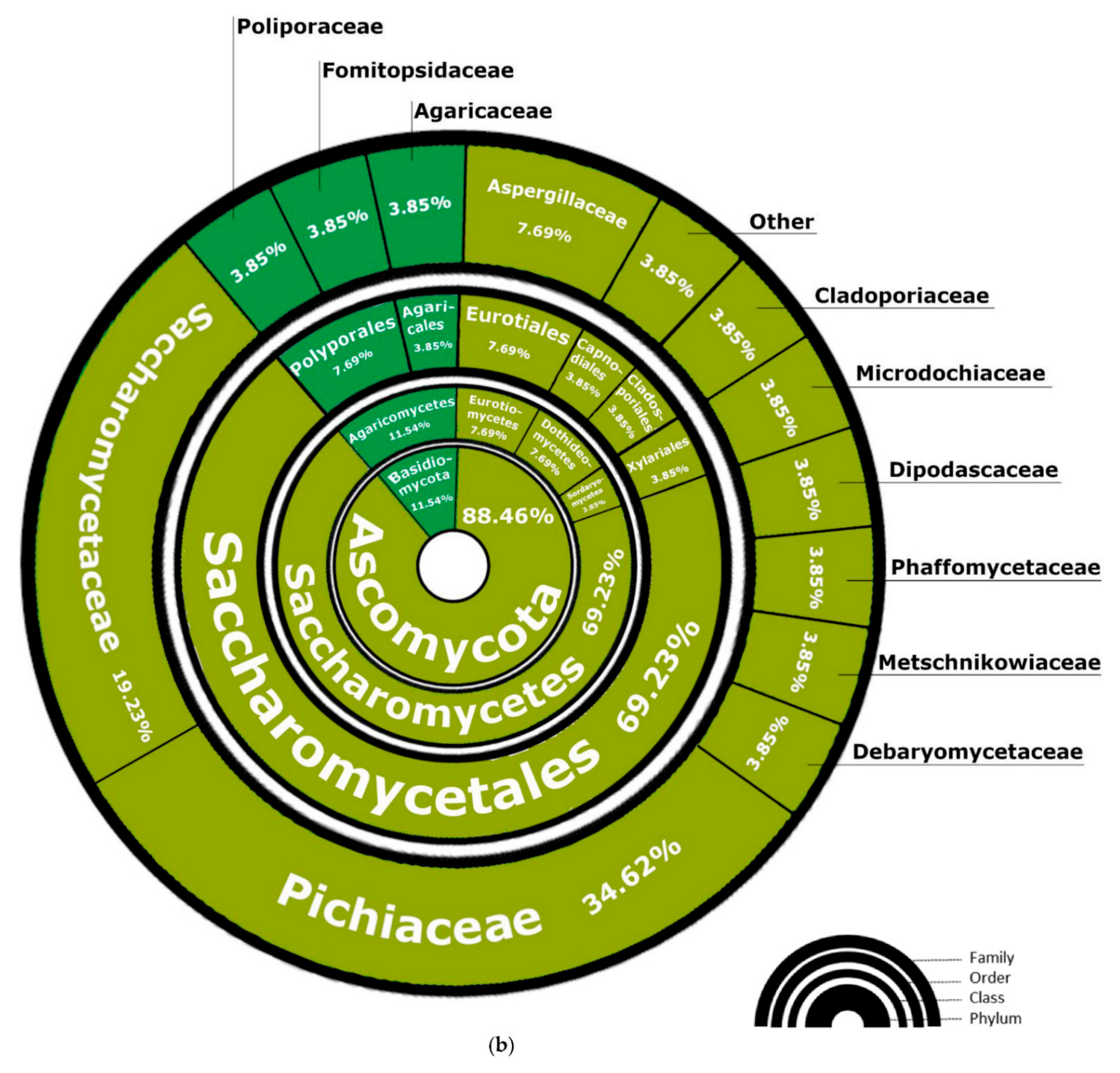
3.2. Fungi—ITS2 Region Analysis
3.2.1. Fungal Composition
3.2.2. Phylogenetic Analysis
4. Discussion
4.1. Exploring the Microbiomes of Mixed-Fermentation Beers
4.2. Lactobacillus spp. and Pediococcus spp. Are Often Identified in Mixed-Fermentation Beers
4.3. Bacteria of the Enterobacteriaceae Family and Their Presence in Spontaneous and Non-Spontaneous Fermentations
4.4. Other Bacteria
4.5. Fungi besides the Brettanomyces spp. and Saccharomyces spp.
4.6. The Importance of Traditional Yeasts in Mixed-Fermentation Beers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

References
- De Roos, J.; De Vuyst, L. Microbial acidification, alcoholization, and aroma production during spontaneous lambic beer production. J. Sci. Food Agric. 2019, 99, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vriesekoop, F.; Krahl, M.; Hucker, B.; Menz, G. 125th Anniversary review: Bacteria in brewing: The good, the bad and the ugly. J. Inst. Brew. 2012, 118, 335–345. [Google Scholar] [CrossRef]
- De Roos, J.; Van der Veken, D.; De Vuyst, L. The interior surfaces of wooden barrels are an additional microbial inoculation source for lambic beer production. Appl. Environ. Microbiol. 2019, 85, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Bongaerts, D.; De Roos, J.; De Vuyst, L. Technological and environmental features determine the uniqueness of the lambic beer microbiota and production process. Appl. Environ. Microbiol. 2021, 87. [Google Scholar] [CrossRef] [PubMed]
- Shayevitz, A.; Harrison, K.; Curtin, C.D. Barrel-Induced Variation in the Microbiome and Mycobiome of Aged Sour Ale and Imperial Porter Beer. J. Am. Soc. Brew. Chem. 2020, 79, 33–40. [Google Scholar] [CrossRef]
- Faria-Oliveira, F.; Diniz, R.H.S.; Godoy-Santos, F.; Piló, F.B.; Mezadri, H.; Castro, I.M.; Brandão, R.L. The Role of Yeast and Lactic Acid Bacteria in the Production of Fermented Beverages in South America. In Food Production and Industry; InTech: Roseville, CA, USA, 2015; Volume i, p. 13. [Google Scholar]
- Spitaels, F.; Wieme, A.D.; Janssens, M.; Aerts, M.; Daniel, H.M.; Van Landschoot, A.; De Vuyst, L.; Vandamme, P. The microbial diversity of traditional spontaneously fermented lambic beer. PLoS ONE 2014, 9, e95384. [Google Scholar] [CrossRef]
- Dysvik, A.; La Rosa, S.L.; De Rouck, G.; Rukke, E.-O.; Westereng, B.; Wicklund, T. Microbial Dynamics in Traditional and Modern Sour Beer Production. Appl. Environ. Microbiol. 2020, 86, 1–14. [Google Scholar] [CrossRef]
- Bossaert, S.; Winne, V.; Van Opstaele, F.; Buyse, J.; Verreth, C.; Herrera-Malaver, B.; Van Geel, M.; Verstrepen, K.J.; Crauwels, S.; De Rouck, G.; et al. Description of the temporal dynamics in microbial community composition and beer chemistry in sour beer production via barrel ageing of finished beers. Int. J. Food Microbiol. 2021, 339, 109030. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Bamforth, C.W.; Mills, D.A. Brewhouse-resident microbiota are responsible for multi-stage fermentation of American coolship ale. PLoS ONE 2012, 7, e35507. [Google Scholar] [CrossRef]
- Witrick, K.; Pitts, E.R.; O’Keefe, S.F. Analysis of Lambic Beer Volatiles during Aging Using Gas Chromatography–Mass Spectrometry (GCMS) and Gas Chromatography–Olfactometry (GCO). Beverages 2020, 6, 31. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tyakht, A.; Kopeliovich, A.; Klimenko, N.; Efimova, D.; Dovidchenko, N.; Odintsova, V.; Kleimenov, M.; Toshchakov, S.; Popova, A.; Khomyakova, M.; et al. Characteristics of bacterial and yeast microbiomes in spontaneous and mixed-fermentation beer and cider. Food Microbiol. 2021, 94, 103658. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Bergsveinson, J.; Ziola, B.; Mills, D.A. Mapping microbial ecosystems and spoilage-gene flow in breweries highlights patterns of contamination and resistance. Elife 2015, 4, e04634. [Google Scholar] [CrossRef]
- Kajala, I.; Bergsveinson, J.; Friesen, V.; Redekop, A.; Juvonen, R.; Storgårds, E.; Ziola, B. Lactobacillus backii and Pediococcus damnosus isolated from 170-year-old beer recovered from a shipwreck lack the metabolic activities required to grow in modern lager beer. FEMS Microbiol. Ecol. 2018, 94, 1–10. [Google Scholar] [CrossRef]
- Rodhouse, L.; Carbonero, F. Overview of craft brewing specificities and potentially associated microbiota. Crit. Rev. Food Sci. Nutr. 2019, 59, 462–473. [Google Scholar] [CrossRef]
- Molinet, J.; Cubillos, F.A. Wild Yeast for the Future: Exploring the Use of Wild Strains for Wine and Beer Fermentation. Front. Genet. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Dysvik, A.; Liland, K.H.; Myhrer, K.S.; Westereng, B.; Rukke, E.O.; de Rouck, G.; Wicklund, T. Pre-fermentation with lactic acid bacteria in sour beer production. J. Inst. Brew. 2019, 125, 342–356. [Google Scholar] [CrossRef]
- Sakamoto, K.; Konings, W.N. Beer spoilage bacteria and hop resistance. Int. J. Food Microbiol. 2003, 89, 105–124. [Google Scholar] [CrossRef]
- Garcia-Garcia, J.H.; Damas-Buenrostro, L.C.; Cabada-Amaya, J.C.; Elias-Santos, M.; Pereyra-Alférez, B. Pediococcus damnosus strains isolated from a brewery environment carry the horA gene. J. Inst. Brew. 2017, 123, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Garcia, J.H.; Galán-Wong, L.J.; Pereyra-Alférez, B.; Damas-Buenrostro, L.C.; Pérez, E.; Carlos Cabada, J. Distribution of lactobacillus and pediococcus in a brewery environment. J. Am. Soc. Brew. Chem. 2017, 75, 312–317. [Google Scholar] [CrossRef]
- Bossaert, S.; Crauwels, S.; Lievens, B.; De Rouck, G. The power of sour—A review: Old traditions, new opportunities. BrewingScience 2019, 72, 78–88. [Google Scholar] [CrossRef]
- Ashtavinayak, P.; Elizabeth, H.A. Review: Gram Negative Bacteria in Brewing. Adv. Microbiol. 2016, 06, 195–209. [Google Scholar] [CrossRef] [Green Version]
- Rodhouse, L. Phylogenetic Diversity of the Bacterial Communities in Craft Beer; University of Arkansas: Fayetteville, AR, USA, 2017. [Google Scholar]
- Justé, A.; Malfliet, S.; Lenaerts, M.; De Cooman, L.; Aerts, G.; Willems, K.A.; Lievens, B. Microflora during malting of barley: Overview and impact on malt quality. BrewingScience 2011, 64, 22–31. [Google Scholar]
- Takahashi, M.; Kita, Y.; Kusaka, K.; Mizuno, A.; Goto-Yamamoto, N. Evaluation of microbial diversity in the pilot-scale beer brewing process by culture-dependent and culture-independent method. J. Appl. Microbiol. 2015, 118, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, Q.; Zhu, S.; Du, F.; Mao, R.; Liu, L.; Tian, B.; Zhu, Y. Biodiversity of non-Saccharomyces yeasts associated with spontaneous fermentation of Cabernet Sauvignon wines from Shangri-La wine region, China. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Van Rijswijck, I.M.H.; Wolkers-Rooijackers, J.C.M.; Abee, T.; Smid, E.J. Performance of non-conventional yeasts in co-culture with brewers’ yeast for steering ethanol and aroma production. Microb. Biotechnol. 2017, 10, 1591–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osburn, K.; Amaral, J.; Metcalf, S.R.; Nickens, D.M.; Rogers, C.M.; Sausen, C.; Caputo, R.; Miller, J.; Li, H.; Tennessen, J.M.; et al. Primary souring: A novel bacteria-free method for sour beer production. Food Microbiol. 2018, 70, 76–84. [Google Scholar] [CrossRef]
- Capece, A.; Romaniello, R.; Siesto, G.; Romano, P. Conventional and Non-Conventional Yeasts in Beer Production. Fermentation 2018, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Zdaniewicz, M.; Satora, P.; Pater, A.; Bogacz, S. Low lactic acid-producing strain of lachancea thermotolerans as a new starter for beer production. Biomolecules 2020, 10, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cason, E.D.; Mahlomaholo, B.J.; Taole, M.M.; Abong, G.O.; Vermeulen, J.G.; de Smidt, O.; Vermeulen, M.; Steyn, L.; Valverde, A.; Viljoen, B. Bacterial and Fungal Dynamics During the Fermentation Process of Sesotho, a Traditional Beer of Southern Africa. Front. Microbiol. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Vogt, E.I.; Kupfer, V.M.; Vogel, R.F.; Niessen, L. Evidence of gushing induction by Penicillium oxalicum proteins. J. Appl. Microbiol. 2017, 122, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Nigam, D.; Asthana, M.; Kumar, A. Penicillium: A fungus in the wine and beer industries; Elsevier B.V.: Amsterdam, The Netherlands, 2017; ISBN 9780444635013. [Google Scholar]
- Serra Colomer, M.; Funch, B.; Forster, J. The raise of Brettanomyces yeast species for beer production. Curr. Opin. Biotechnol. 2019, 56, 30–35. [Google Scholar] [CrossRef]
- Sobel, J.; Henry, L.; Rotman, N.; Rando, G. BeerDeCoded: The open beer metagenome project. F1000Research 2017, 6, 1676. [Google Scholar] [CrossRef] [PubMed]
- Colomer, M.S.; Chailyan, A.; Fennessy, R.T.; Olsson, K.F.; Johnsen, L.; Solodovnikova, N.; Forster, J. Assessing Population Diversity of Brettanomyces Yeast Species and Identification of Strains for Brewing Applications. Front. Microbiol. 2020, 11, 1–21. [Google Scholar] [CrossRef]
- Stewart, G. Saccharomyces species in the Production of Beer. Beverages 2016, 2, 34. [Google Scholar] [CrossRef]
- Lengeler, K.B.; Stovicek, V.; Fennessy, R.T.; Katz, M.; Förster, J. Never Change a Brewing Yeast? Why Not, There Are Plenty to Choose From. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Gallone, B.; Steensels, J.; Mertens, S.; Dzialo, M.C.; Gordon, J.L.; Wauters, R.; Theßeling, F.A.; Bellinazzo, F.; Saels, V.; Herrera-Malaver, B.; et al. Interspecific hybridization facilitates niche adaptation in beer yeast. Nat. Ecol. Evol. 2019, 3, 1562–1575. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Origin | Material | Spontaneous Fermentation | Fruits, Woods, Flowers, or Another Microbe Source Added | Culture Maintenance Time | Commercial Strains Inoculated |
|---|---|---|---|---|---|---|
| 1 | Jerusalem (IL*) | Culture pre-pitch | YES | NO | 6–12 months | - |
| 2 | Alberta (CA) | Beer/slurry | NO | YES | 1–6 months | New W. Saison (Escarpment Labs) and Brett ‘M’ (Escarpment Labs) |
| 3 | Alberta (CA) | Beer | YES | YES | 6–12 months | - |
| 4 | Alberta (CA) | Beer | YES | NO | 6–12 months | - |
| 5 | Alberta (CA) | Beer | YES | YES | 6–12 months | - |
| 6 | Alberta (CA) | Beer | YES | YES | 6–12 months | - |
| 7 | Alberta (CA) | Beer | YES | YES | 6–12 months | - |
| 8 | Alberta (CA) | Beer | YES | YES | 6–12 months | - |
| 9 | Washington (US) | Culture pre-pitch | YES | NO | 2–3 years | - |
| 10 | Washington (US) | Culture pre-pitch | YES | NO | 6–12 months | - |
| 11 | Ohio (US) | Beer/slurry | NO | NO | 6–12 months | Sour Solera (Bootleg Biology); Mélange (The Yeast Bay); BugCounty (East Coast Yeast); and dregs from beer bottles |
| 12 | Nevada (US) | Beer | NO | YES | 6–12 months | Dregs from beer bottles |
| 13 | Nevada (US) | Beer | NO | YES | 6–12 months | Dregs from beer bottles |
| 14 | California (US) | Beer | NO | NO | 6–12 months | in-house culture |
| 15 | California (US) | Culture pre-pitch | YES | NO | 5–6 years | - |
| 16 | California (US) | Beer | NO | NO | 4–5 years | in-house culture |
| 17 | California (US) | Beer | YES | NO | 1–6 months | - |
| 18 | California (US) | Beer | NO | NO | 2–3 years | WY3763 (Wyeast); WY3711 (Wyeast); and WLP650 (White Labs) |
| 19 | California (US) | Beer | NO | NO | 1–2 years | WLP565 (White Labs) and dregs from beer bottles |
| 20 | Michigan (US) | Beer | YES | NO | 1–6 months | CBC-1 (Lallemand) for bottling |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piraine, R.E.A.; Leite, F.P.L.; Bochman, M.L. Mixed-Culture Metagenomics of the Microbes Making Sour Beer. Fermentation 2021, 7, 174. https://doi.org/10.3390/fermentation7030174
Piraine REA, Leite FPL, Bochman ML. Mixed-Culture Metagenomics of the Microbes Making Sour Beer. Fermentation. 2021; 7(3):174. https://doi.org/10.3390/fermentation7030174
Chicago/Turabian StylePiraine, Renan Eugênio Araujo, Fábio Pereira Leivas Leite, and Matthew L. Bochman. 2021. "Mixed-Culture Metagenomics of the Microbes Making Sour Beer" Fermentation 7, no. 3: 174. https://doi.org/10.3390/fermentation7030174
APA StylePiraine, R. E. A., Leite, F. P. L., & Bochman, M. L. (2021). Mixed-Culture Metagenomics of the Microbes Making Sour Beer. Fermentation, 7(3), 174. https://doi.org/10.3390/fermentation7030174