
Predictive Microbial Community and Functional Gene Expression Profiles in Pineapple Peel Fermentation Using 16S rRNA Gene Sequences

 ,
,  , ,
, ,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Fermentation
2.2. Analysis of Ethanol Concentration
2.3. Metagenomic Analysis
2.3.1. Total Genomic DNA Extraction
2.3.2. Amplicon and Libraries Generation
2.3.3. Sequencing Data Processing
2.3.4. OTU Cluster and Taxonomic Annotation
2.3.5. Alpha Diversity
2.3.6. Beta Diversity
2.4. PiCRUSt Analysis
3. Results
3.1. Ethanol Concentration
3.2. Bacterial Community Profile in the Pineapple Peel Fermentation
3.2.1. Relative Abundance
3.2.2. Taxonomic Abundance Cluster Heatmap
3.2.3. Alpha Diversity
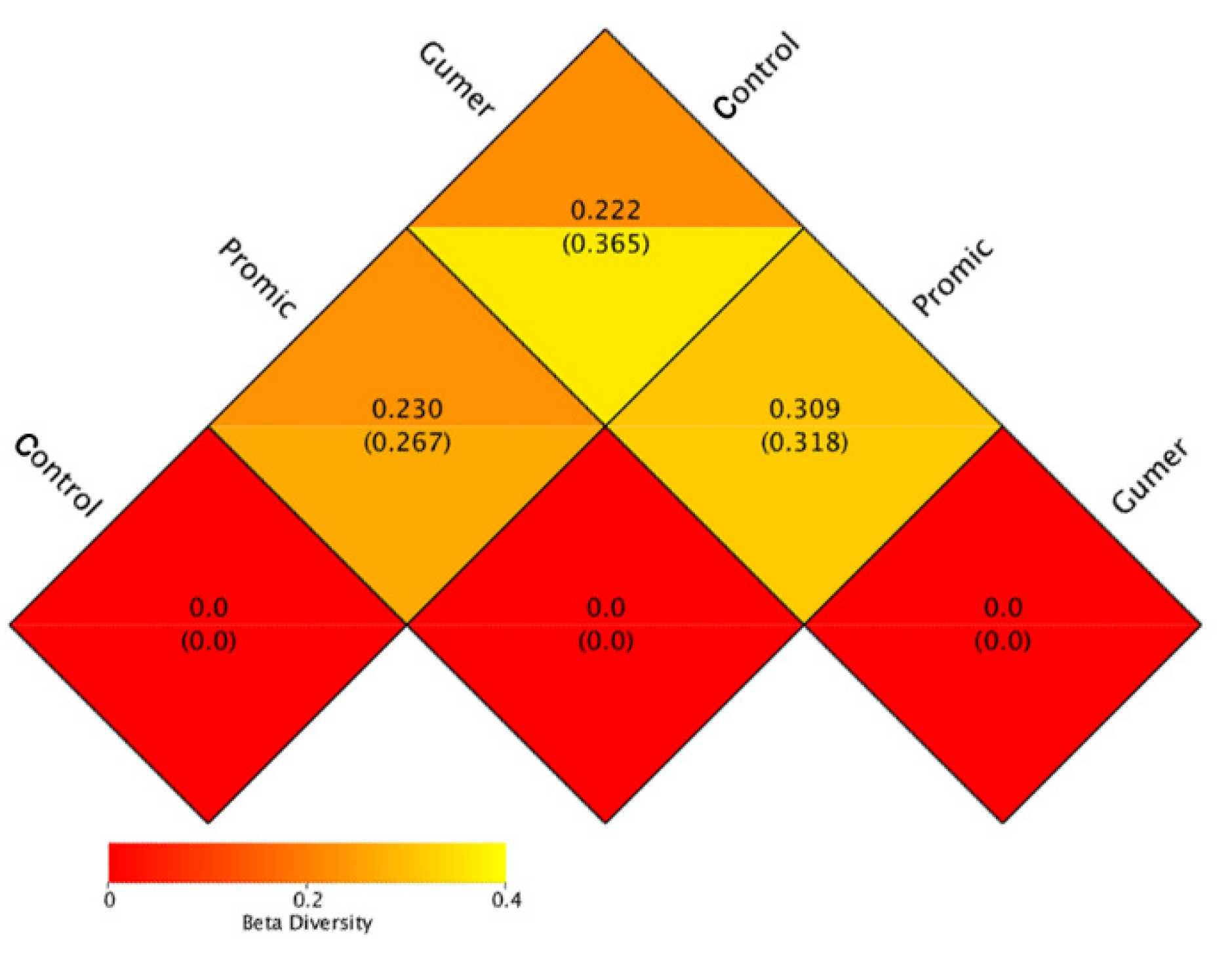
3.2.4. Beta Diversity
3.2.5. Phylogenetic Tree of the Phyla
3.2.6. Principal Coordinates Analysis
3.3. KEGG-Based PICRUSt Analysis
3.3.1. Prediction of Functional Genes Expression
3.3.2. Prediction of Microbial Metabolism
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amar, A.T.; Tong, P.S.; Ng, C.K. The MD2 “Super Sweet” pineapple (Ananas comosus). Utar Agric. Sci. J. 2015, 1, 14–16. [Google Scholar]
- Saraswaty, V.; Risdian, C.; Primadona, I.; Andriyani, R.; Andayani, D.G.S.; Mozef, T. Pineapple Peel Wastes as a Potential Source of Antioxidant Compounds. In IOP Conference Series: Earth and Environmental Science; IOP Publishing: Bristol, UK, 2017; Volume 60, p. 12013. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-L.; Chow, C.J.; Fang, Y. Preparation and physicochemical properties of fiber-rich fraction from pineapple peels as a potential ingredient. J. Food Drug Anal. 2011, 19, 318–323. [Google Scholar] [CrossRef]
- Li, T.; Shen, P.; Liu, W.; Liu, C.; Liang, R.; Yan, N.; Chen, J. Major polyphenolics in pineapple peels and their antioxidant interactions. Int. J. Food Prop. 2014, 17, 1805–1817. [Google Scholar] [CrossRef]
- Lubaina, A.S.; Renjith, P.; Roshni, A. Identification and quantification of polyphenols from pineapple peel by high performance liquid chromatography analysis. Adv. Zool. Bot. 2020, 8, 431–438. [Google Scholar] [CrossRef]
- Saleh, M.; Amro, L.; Barakat, H.; Baker, R.; Reyash, A.A.; Amro, R.; Qasem, J. Fruit by-product processing and bioactive compounds. J. Food Qual. 2021, 2021, 5513358. [Google Scholar] [CrossRef]
- Boo, Y.C. Can plant phenolic compounds protect the skin from airborne particulate matter? Antioxidants 2019, 8, 379. [Google Scholar] [CrossRef] [Green Version]
- Pawlowska, E.; Szczepanska, J.; Koskela, A.; Kaarniranta, K.; Blasiak, J. Dietary polyphenols in age-related macular degeneration: Protection against oxidative stress and beyond. Oxid. Med. Cell. Longev. 2019, 2019, 9682318. [Google Scholar] [CrossRef]
- Tallei, T.E.; Marfuah, S.; Abas, A.H.; Abram, A.A.D.P.; Pasappa, N.; Anggini, P.S.; Soegoto, A.S.; Fatimawali; Emran, T.B. Nata as a source of dietary fiber with numerous health benefits. J. Adv. Biotechnol. Exp. Ther. 2022, 5, 189–197. [Google Scholar] [CrossRef]
- Sharma, R.; Garg, P.; Kumar, P.; Bhatia, S.K.; Kulshrestha, S. Microbial fermentation and its role in quality improvement of fermented foods. Fermentation 2020, 6, 106. [Google Scholar] [CrossRef]
- Cichońska, P.; Ziarno, M. Legumes and legume-based beverages fermented with lactic acid bacteria as a potential carrier of probiotics and prebiotics. Microorganisms 2022, 10, 91. [Google Scholar] [CrossRef]
- Rezac, S.; Kok, C.R.; Heermann, M.; Hutkins, R. Fermented foods as a dietary source of live organisms. Front. Microbiol. 2018, 9, 1785. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhao, Z.; Wu, X.; Lin, W.; Qin, Y.; Chen, H.; Wan, Y.; Zhou, C.; Bu, T.; Chen, H.; et al. A Review on fruit and vegetable fermented beverage-benefits of microbes and beneficial effects. Food Rev. Int. 2022, 1–38. [Google Scholar] [CrossRef]
- Agyirifo, D.S.; Wamalwa, M.; Otwe, E.P.; Galyuon, I.; Runo, S.; Takrama, J.; Ngeranwa, J. Metagenomics analysis of cocoa bean fermentation microbiome identifying species diversity and putative functional capabilities. Heliyon 2019, 5, e02170. [Google Scholar] [CrossRef] [Green Version]
- Fasesan, D.; Dawkins, K.; Ramirez, R.; Rasheed-Jada, H.; Onilude, A.; Nash, O.; Esiobu, N. Analysis of a tropical warm spring microbiota using 16S rRNA metabarcoding. Adv. Microbiol. 2020, 10, 145–165. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Sun-Waterhouse, D.; Waterhouse, G.I.N.; Cui, C.; Ruan, Z. Fermentation-enabled wellness foods: A fresh perspective. Food Sci. Hum. Wellness 2019, 8, 203–243. [Google Scholar] [CrossRef]
- Amit, S.K.; Uddin, M.M.; Rahman, R.; Islam, S.M.R.; Khan, M.S. A review on mechanisms and commercial aspects of food preservation and processing. Agric. Food Secur. 2017, 6, 51. [Google Scholar] [CrossRef]
- Romero-Luna, H.E.; Hernández-Sánchez, H.; Dávila-Ortiz, G. Traditional fermented beverages from Mexico as a potential probiotic source. Ann. Microbiol. 2017, 67, 577–586. [Google Scholar] [CrossRef]
- Kergourlay, G.; Taminiau, B.; Daube, G.; Champomier Vergès, M.-C. Metagenomic insights into the dynamics of microbial communities in food. Int. J. Food Microbiol. 2015, 213, 31–39. [Google Scholar] [CrossRef]
- Arıkan, M.; Mitchell, A.L.; Finn, R.D.; Gürel, F. Microbial composition of Kombucha determined using amplicon sequencing and shotgun metagenomics. J. Food Sci. 2020, 85, 455–464. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Huo, D.; Li, W.; Hu, Q.; Xu, C.; Liu, S.; Li, C. Metagenomic approach reveals microbial diversity and predictive microbial metabolic pathways in Yucha, a traditional Li fermented food. Sci. Rep. 2016, 6, 32524. [Google Scholar] [CrossRef]
- Butnariu, M.; Butu, A. 10—The Evolution and the Development Phases of Wine. In Alcoholic Beverages; Grumezescu, A.M., Holban, A.M., Eds.; Woodhead Publishing: Sawston, UK, 2019; pp. 303–345. ISBN 978-0-12-815269-0. [Google Scholar]
- Pauzi, N.; Man, S.; Nawawi, M.S.A.M.; Abu-Hussin, M.F. Ethanol standard in halal dietary product among Southeast Asian halal governing bodies. Trends Food Sci. Technol. 2019, 86, 375–380. [Google Scholar] [CrossRef]
- Yang, X.; Hu, W.; Xiu, Z.; Jiang, A.; Yang, X.; Saren, G.; Ji, Y.; Guan, Y.; Feng, K. Microbial community dynamics and metabolome changes during spontaneous fermentation of Northeast sauerkraut from different households. Front. Microbiol. 2020, 11, 1878. [Google Scholar] [CrossRef]
- Shangpliang, H.N.J.; Rai, R.; Keisam, S.; Jeyaram, K.; Tamang, J.P. Bacterial community in naturally fermented milk products of Arunachal Pradesh and Sikkim of India analysed by high-throughput amplicon sequencing. Sci. Rep. 2018, 8, 1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radita, R.; Suwanto, A.; Kurosawa, N.; Wahyudi, A.T.; Rusmana, I. Firmicutes is the predominant bacteria in tempeh. Int. Food Res. J. 2018, 25, 2313–2320. [Google Scholar]
- Zhao, X.; Xiang, F.; Tang, F.; Cai, W.; Guo, Z.; Hou, Q.; Yang, X.; Song, W.; Shan, C. Bacterial communities and prediction of microbial metabolic pathway in rice wine Koji from different regions in China. Front. Microbiol. 2022, 12, 748779. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, Y.; Cai, W.; Yang, M.; Zhong, X.; Guo, Z.; Shan, C. High-throughput sequencing-based analysis of microbial diversity in rice wine Koji from different areas. Curr. Microbiol. 2020, 77, 882–889. [Google Scholar] [CrossRef]
- Guarner, F.; Malagelada, J.-R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Beaugerie, L.; Petit, J.-C. Antibiotic-associated diarrhoea. Best Pract. Res. Clin. Gastroenterol. 2004, 18, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Lonvaud-Funel, A. Leuconostocaceae Family, 2nd ed.; Batt, C.A., Tortorello, M.L.B.T.-E., Eds.; Academic Press: Oxford, UK, 2014; pp. 455–465. ISBN 978-0-12-384733-1. [Google Scholar]
- Adewumi, G.A. Health-Promoting Fermented Foods. In Encyclopedia of Food Chemistry; Melton, L., Shahidi, F., Varelis, P., Eds.; Academic Press: Oxford, UK, 2019; pp. 399–418. ISBN 978-0-12-814045-1. [Google Scholar]
- De Bruyne, K.; Camu, N.; Lefebvre, K.; De Vuyst, L.; Vandamme, P. Weissella ghanensis sp. nov., isolated from a Ghanaian cocoa fermentation. Int. J. Syst. Evol. Microbiol. 2008, 58, 2721–2725. [Google Scholar] [CrossRef]
- Delavenne, E.; Cliquet, S.; Trunet, C.; Barbier, G.; Mounier, J.; Le Blay, G. Characterization of the antifungal activity of Lactobacillus harbinensis K.V9.3.1Np and Lactobacillus rhamnosus K.C8.3.1I in yogurt. Food Microbiol. 2015, 45, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Nethery, M.A.; Henriksen, E.D.; Daughtry, K.V.; Johanningsmeier, S.D.; Barrangou, R. Comparative genomics of eight Lactobacillus buchneri strains isolated from food spoilage. BMC Genom. 2019, 20, 902. [Google Scholar] [CrossRef]
- Duranti, S.; Ruiz, L.; Lugli, G.A.; Tames, H.; Milani, C.; Mancabelli, L.; Mancino, W.; Longhi, G.; Carnevali, L.; Sgoifo, A.; et al. Bifidobacterium adolescentis as a key member of the human gut microbiota in the production of GABA. Sci. Rep. 2020, 10, 14112. [Google Scholar] [CrossRef] [PubMed]
- Tamang, J.P.; Shin, D.-H.; Jung, S.-J.; Chae, S.-W. Functional properties of microorganisms in fermented foods. Front. Microbiol. 2016, 7, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, B.; Chen, J.-S.; Hsu, B.-M.; Chu, I.-T.; Koner, S.; Chen, T.-H.; Rathod, J.; Chan, M.W.Y. Deciphering bacterial community structure, functional prediction and food safety assessment in fermented fruits using next-generation 16S rRNA amplicon sequencing. Microorganisms 2021, 9, 1574. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Lv, M.; Shao, Z.; Hungwe, M.; Wang, J.; Bai, X.; Xie, J.; Wang, Y.; Geng, W. Metabolism characteristics of lactic acid bacteria and the expanding applications in food industry. Front. Bioeng. Biotechnol. 2021, 9, 612285. [Google Scholar] [CrossRef] [PubMed]
- Fusco, V.; Quero, G.M.; Cho, G.-S.; Kabisch, J.; Meske, D.; Neve, H.; Bockelmann, W.; Franz, C.M.A.P. The genus Weissella: Taxonomy, ecology and biotechnological potential. Front. Microbiol. 2015, 6, 155. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.; Lu, M.; Park, C.; Park, C.; bin Oh, H.; Lee, S.Y.; Lee, J. Dynamic modeling of lactic acid fermentation metabolism with Lactococcus lactis. J. Microbiol. Biotechnol. 2011, 21, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.E.; Chun, B.H.; Kim, K.H.; Park, D.; Roh, S.W.; Lee, S.H.; Jeon, C.O. Genomic and metatranscriptomic analyses of Weissella koreensis reveal its metabolic and fermentative features during kimchi fermentation. Food Microbiol. 2018, 76, 1–10. [Google Scholar] [CrossRef]
- Xie, M.; An, F.; Zhao, Y.; Wu, R.; Wu, J. Metagenomic analysis of bacterial community structure and functions during the fermentation of da-jiang, a Chinese traditional fermented food. LWT 2020, 129, 109450. [Google Scholar] [CrossRef]
- Wu, J.; Tian, T.; Liu, Y.; Shi, Y.; Tao, D.; Wu, R.; Yue, X. The dynamic changes of chemical components and microbiota during the natural fermentation process in Da-Jiang, a Chinese popular traditional fermented condiment. Food Res. Int. 2018, 112, 457–467. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Average Concentration of EtOH (%) |
|---|---|
| Control | 0.0838 |
| Promic | 0.0661 |
| Gumer | 0.0846 |
| Sample Name | Observed Species | Shannon | Simpson | Chao1 | ACE |
|---|---|---|---|---|---|
| Control | 96 | 1.851 | 0.609 | 100.091 | 100.091 |
| Promic | 97 | 1.794 | 0.530 | 104.583 | 104.583 |
| Gumer | 104 | 0.408 | 0.080 | 108.200 | 114.441 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tallei, T.E.; Fatimawali; Yelnetty, A.; Kusumawaty, D.; Effendi, Y.; Park, M.N.; Alhumaydhi, F.A.; Emran, T.B.; Kim, B. Predictive Microbial Community and Functional Gene Expression Profiles in Pineapple Peel Fermentation Using 16S rRNA Gene Sequences. Fermentation 2022, 8, 194. https://doi.org/10.3390/fermentation8050194
Tallei TE, Fatimawali, Yelnetty A, Kusumawaty D, Effendi Y, Park MN, Alhumaydhi FA, Emran TB, Kim B. Predictive Microbial Community and Functional Gene Expression Profiles in Pineapple Peel Fermentation Using 16S rRNA Gene Sequences. Fermentation. 2022; 8(5):194. https://doi.org/10.3390/fermentation8050194
Chicago/Turabian StyleTallei, Trina Ekawati, Fatimawali, Afriza Yelnetty, Diah Kusumawaty, Yunus Effendi, Moon Nyeo Park, Fahad A. Alhumaydhi, Talha Bin Emran, and Bonglee Kim. 2022. "Predictive Microbial Community and Functional Gene Expression Profiles in Pineapple Peel Fermentation Using 16S rRNA Gene Sequences" Fermentation 8, no. 5: 194. https://doi.org/10.3390/fermentation8050194
APA StyleTallei, T. E., Fatimawali, Yelnetty, A., Kusumawaty, D., Effendi, Y., Park, M. N., Alhumaydhi, F. A., Emran, T. B., & Kim, B. (2022). Predictive Microbial Community and Functional Gene Expression Profiles in Pineapple Peel Fermentation Using 16S rRNA Gene Sequences. Fermentation, 8(5), 194. https://doi.org/10.3390/fermentation8050194