
Physiological and Molecular Characterization of Yeast Cultures Pre-Adapted for Fermentation of Lignocellulosic Hydrolysate
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Raw Material and Pretreatment
2.2. Yeast Cultivation
2.3. Enzymatic Activity in Crude Cell Extracts
2.4. Microarray Experiments
2.5. Analysis of Microarray Data
2.6. Identification of Phenolic Compounds
2.7. SSF Experiments
2.8. Yield Calculations
2.9. Biomass and Metabolite Analysis
3. Results
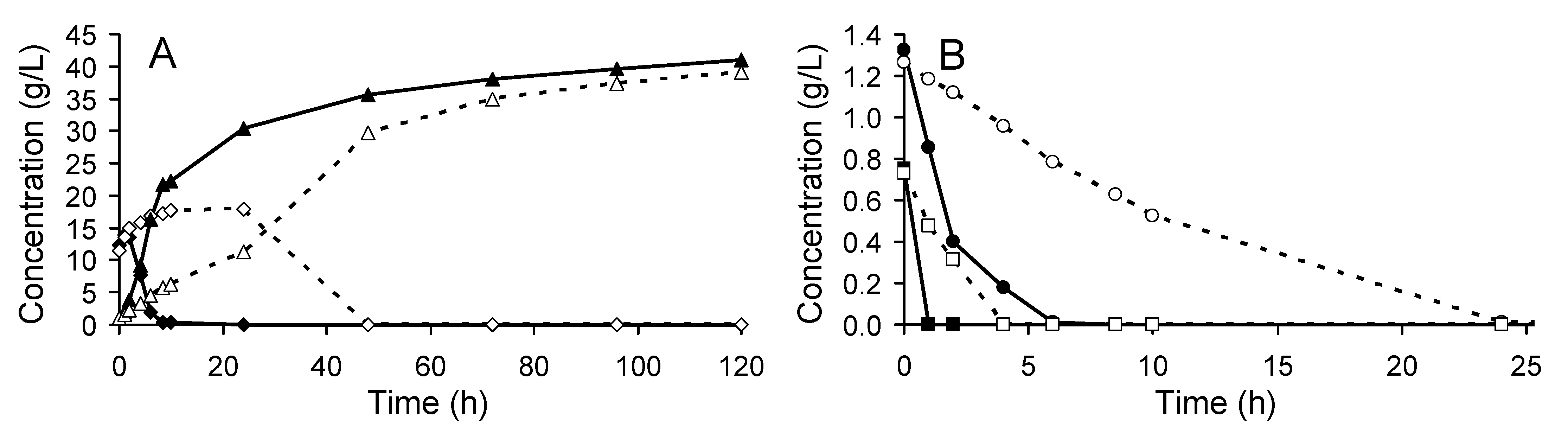
3.1. Yeast Performance in the SSF Experiments
3.2. Effects of Hydrolysate on Yeast Pre-Cultures
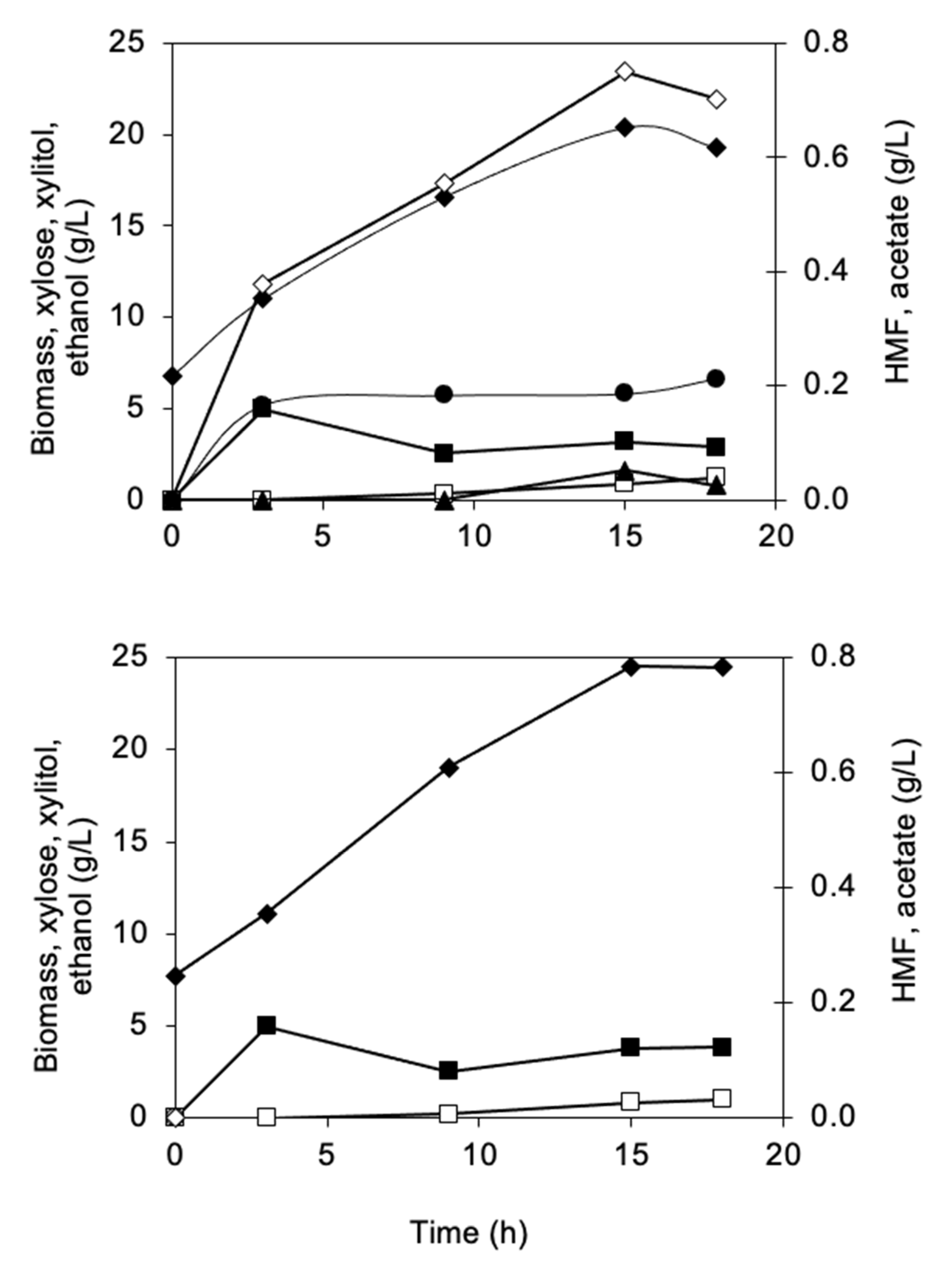
3.2.1. Biomass and Metabolite Analysis
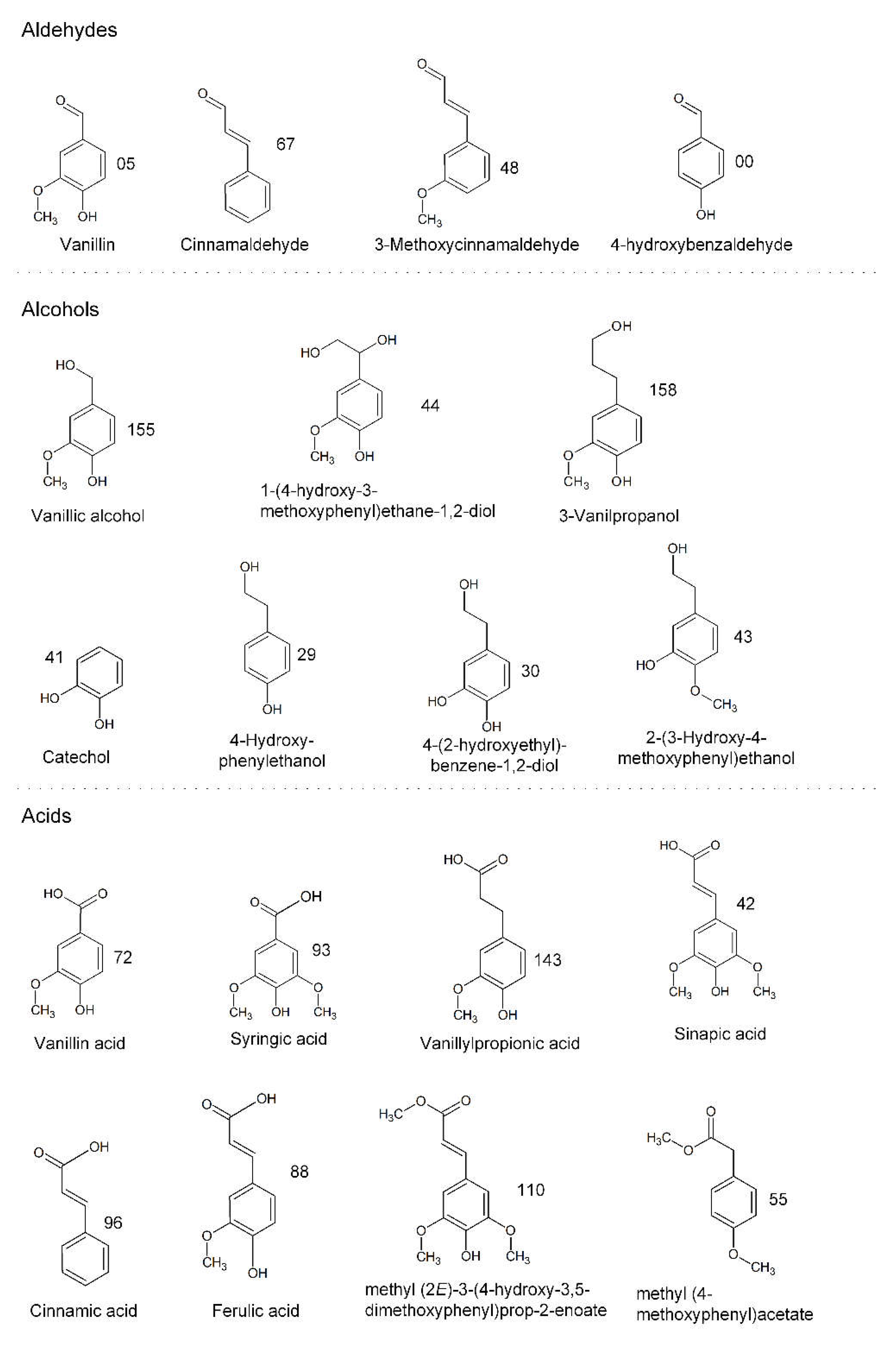
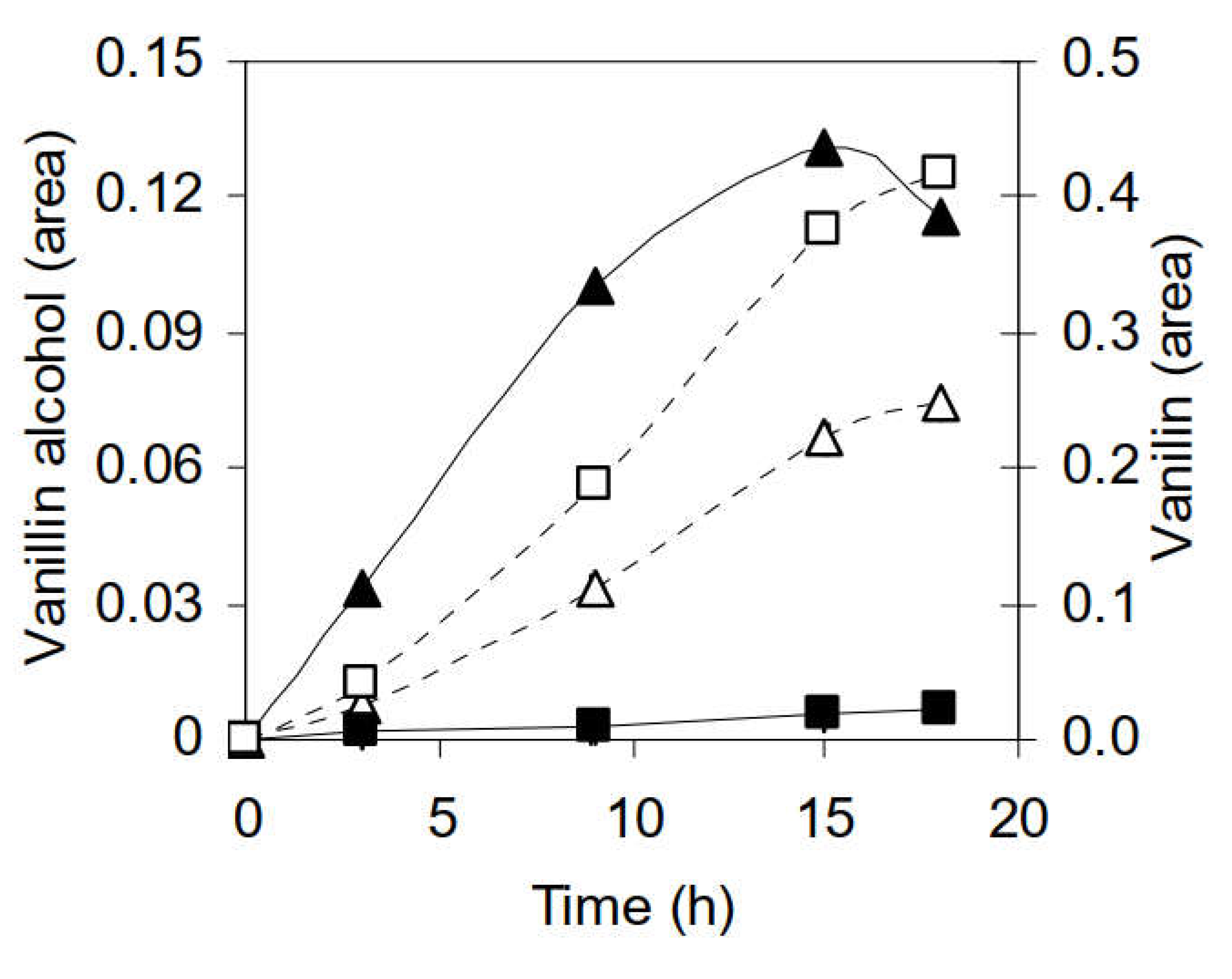
3.2.2. Identification of Phenolic Compounds in the Propagation Step on Spruce Hydrolysate Medium
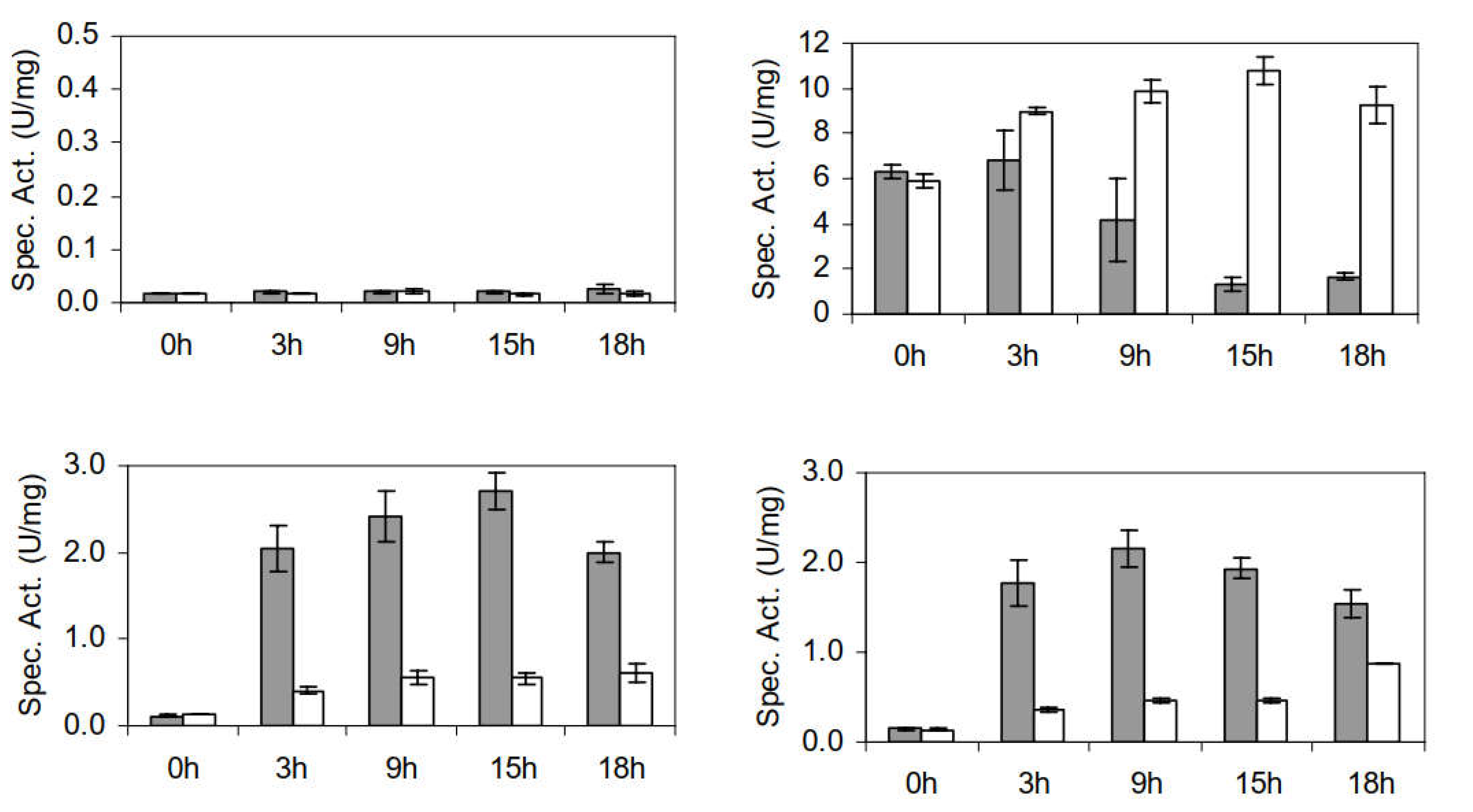
3.2.3. Furaldehyde Reduction Activity
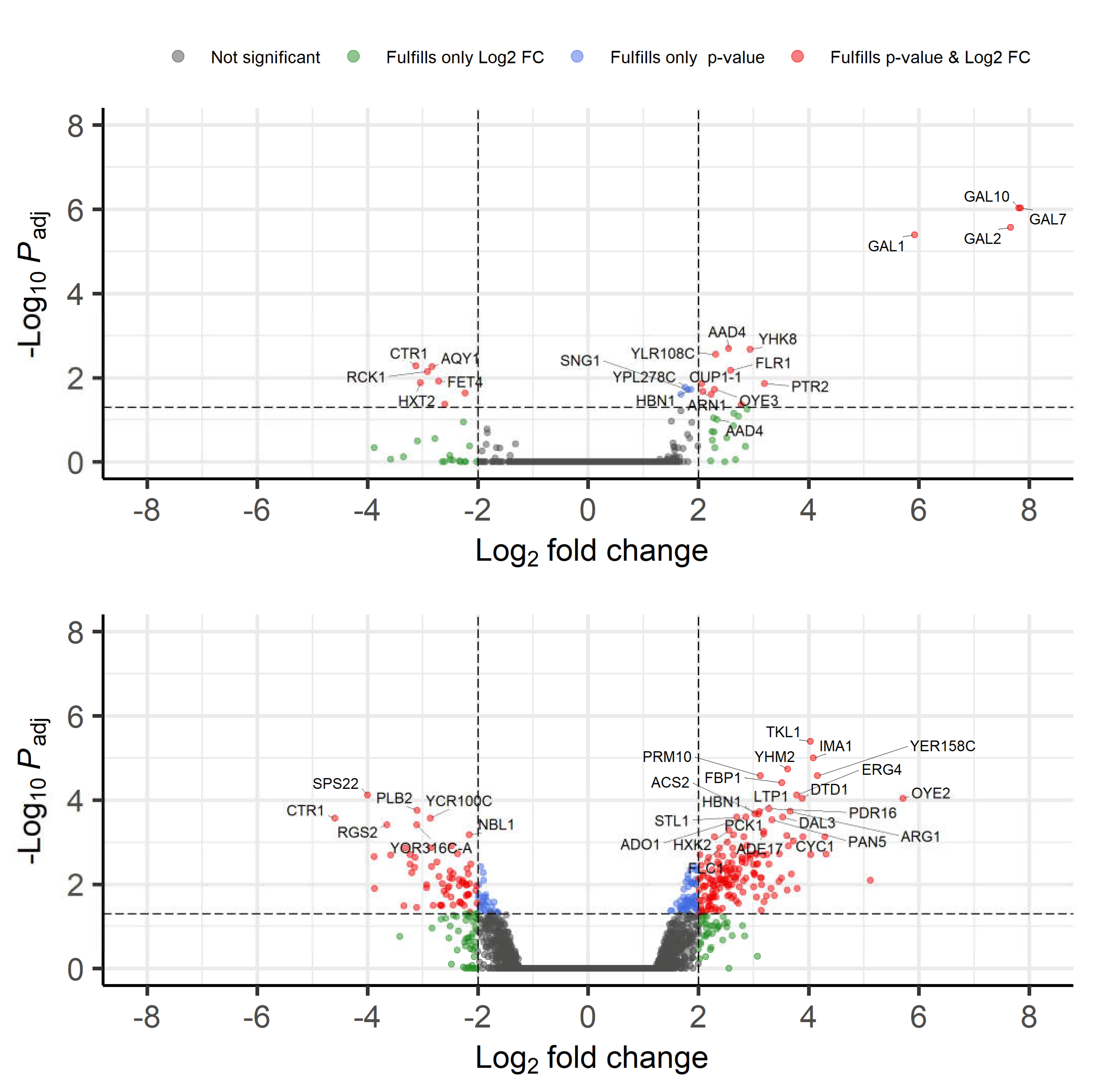
3.2.4. Transcriptome Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galbe, M.; Zacchi, G. Pretreatment of lignocellulosic materials for efficient bioethanol production. Adv. Biochem. Eng. Biotechnol. 2007, 108, 41–65. [Google Scholar] [CrossRef] [PubMed]
- Hahn-Hägerdal, B.; Karhumaa, K.; Jeppsson, M.; Gorwa-Grauslund, M.F. Metabolic engineering for pentose utilization in Saccharomyces cerevisiae. Adv. Biochem. Eng./Biotechnol. 2007, 108, 147–177. [Google Scholar] [CrossRef]
- Almeida, J.R.M.; Modig, T.; Petersson, A.; Hahn-Hägerdal, B.; Lidén, G.; Gorwa-Grauslund, M.F. Increased tolerance and conversion of inhibitors in lignocellulosic hydrolysates by Saccharomyces cerevisiae. J. Chem. Technol. Biotechnol. 2007, 82, 340–349. [Google Scholar] [CrossRef]
- Klinke, H.B.; Thomsen, A.B.; Ahring, B.K. Inhibition of ethanol-producing yeast and bacteria by degradation products produced during pre-treatment of biomass. Appl. Microbiol. Biotechnol. 2004, 66, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Buschke, N.; Schäfer, R.; Becker, J.; Wittmann, C. Metabolic engineering of industrial platform microorganisms for biorefinery applications—Optimization of substrate spectrum and process robustness by rational and evolutive strategies. Bioresour. Technol. 2013, 135, 544–554. [Google Scholar] [CrossRef]
- Hong, K.-K.; Nielsen, J. Metabolic engineering of Saccharomyces cerevisiae: A key cell factory platform for future biorefineries. Cell. Mol. Life Sci. 2012, 69, 2671–2690. [Google Scholar] [CrossRef]
- Brandberg, T.; Franzén, C.J.; Gustafsson, L. The fermentation performance of nine strains of Saccharomyces cerevisiae in batch and fed-batch cultures in dilute-acid wood hydrolysate. J. Biosci. Bioeng. 2004, 98, 122–125. [Google Scholar] [CrossRef]
- Heer, D.; Sauer, U. Identification of furfural as a key toxin in lignocellulosic hydrolysates and evolution of a tolerant yeast strain. Microb. Biotechnol. 2008, 1, 497–506. [Google Scholar] [CrossRef]
- Almeida, J.R.M.; Bertilsson, M.; Gorwa-Grauslund, M.F.; Gorsich, S.; Lidén, G. Metabolic effects of furaldehydes and impacts on biotechnological processes. Appl. Microbiol. Biotechnol. 2009, 82, 625–638. [Google Scholar] [CrossRef]
- Gorsich, S.W.; Dien, B.S.; Nichols, N.N.; Slininger, P.J.; Liu, Z.L.; Skory, C.D. Tolerance to furfural-induced stress is associated with pentose phosphate pathway genes ZWF1, GND1, RPE1, and TKL1 in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2006, 71, 339–349. [Google Scholar] [CrossRef]
- Larsson, S.; Palmqvist, E.; Hahn-Hägerdal, B.; Tengborg, C.; Stenberg, K.; Zacchi, G.; Nilvebrant, N.-O. The generation of fermentation inhibitors during dilute acid hydrolysis of softwood. Enzym. Microb. Technol. 1999, 24, 151–159. [Google Scholar] [CrossRef]
- Guaragnella, N.; Bettiga, M. Acetic acid stress in budding yeast: From molecular mechanisms to applications. Yeast 2021, 38, 391–400. [Google Scholar] [CrossRef]
- Larsson, S.; Quintana-Sáinz, A.; Reimann, A.; Nilvebrant, N.-O.; Jönsson, L. Influence of lignocellulose-derived aromatic compounds on oxygen-limited growth and ethanolic fermentation by Saccharomyces cerevisiae. Appl. Biochem. Biotechnol. 2000, 84–86, 617–632. [Google Scholar] [CrossRef]
- Adeboye, P.T.; Bettiga, M.; Olsson, L. The chemical nature of phenolic compounds determines their toxicity and induces distinct physiological responses in Saccharomyces cerevisiae in lignocellulose hydrolysates. AMB Express 2014, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.L.; Slininger, P.J.; Gorsich, S.W. Enhanced biotransformation of furfural and hydroxymethylfurfural by newly developed ethanologenic yeast strains. Appl. Biochem. Biotechnol. 2005, 121–124, 451–460. [Google Scholar] [CrossRef]
- Martin, C.; Marcet, M.; Almazan, O.; Jönsson, L.J. Adaptation of a recombinant xylose-utilizing Saccharomyces cerevisiae strain to a sugarcane bagasse hydrolysate with high content of fermentation inhibitors. Bioresour. Technol. 2007, 98, 1767–1773. [Google Scholar] [CrossRef]
- Li, B.; Liu, N.; Zhao, X. Response mechanisms of Saccharomyces cerevisiae to the stress factors present in lignocellulose hydrolysate and strategies for constructing robust strains. Biotechnol. Biofuels. Bioprod. 2022, 15, 28. [Google Scholar] [CrossRef]
- Cámara, E.; Olsson, L.; Zrimec, J.; Zelezniak, A.; Geijer, C.; Nygård, Y. Data mining of Saccharomyces cerevisiae mutants engineered for increased tolerance towards inhibitors in lignocellulosic hydrolysates. Biotechnol. Adv. 2022, 57, 107947. [Google Scholar] [CrossRef]
- Alkasrawi, M.; Rudolf, A.; Lidén, G.; Zacchi, G. Influence of strain and cultivation procedure on the performance of simultaneous saccharification and fermentation of steam pretreated spruce. Enzym. Microb. Technol. 2006, 38, 279–286. [Google Scholar] [CrossRef]
- Van Dijk, M.; Rugbjerg, P.; Nygård, Y.; Olsson, L. RNA sequencing reveals metabolic and regulatory changes leading to more robust fermentation performance during short-term adaptation of Saccharomyces cerevisiae to lignocellulosic inhibitors. Biotechnol. Biofuels. 2021, 14, 201. [Google Scholar] [CrossRef]
- Almeida, J.R.M.; Karhumaa, K.; Bengtsson, O.; Gorwa-Grauslund, M.-F. Screening of Saccharomyces cerevisiae strains with respect to anaerobic growth in non-detoxified lignocellulose hydrolysate. Bioresour. Technol. 2009, 100, 3674–3677. [Google Scholar] [CrossRef] [PubMed]
- Modig, T.; Almeida, J.R.M.; Gorwa-Grauslund, M.F.; Lidén, G. Variability of the response of Saccharomyces cerevisiae strains to lignocellulose hydrolysate. Biotechnol. Bioeng. 2008, 100, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Tempelton, D.; Crocker, D. Determination of structural carbohydrates and lignin in biomass. Lab. Anal. Proced. Natl. Renew. Energy Lab. Gold. CO USA 2008, 1617, 1–16. [Google Scholar]
- Ruiz, R.; Ehrman, T. HPLC analysis of liquid fractions of process samples for monomeric sugars and cellobiose. In Laboratory Analytical Procedure (LAP 013); National Renewable Energy Laboratory: Golden, CO, USA, 1996. [Google Scholar]
- Petersson, A.; Almeida, J.R.M.; Modig, T.; Karhumaa, K.; Hahn-Hägerdal, B.; Gorwa-Grauslund, M.F.; Lidén, G. A 5-hydroxymethyl furfural reducing enzyme encoded by the Saccharomyces cerevisiae ADH6 gene conveys HMF tolerance. Yeast 2006, 23, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing: Vienna, Austria. 2022. Available online: https://www.R-project.org/ (accessed on 10 November 2022).
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Irizarry, R.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.; Antonellis, K.; Scherf, U.; Speed, T. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Smyth, G.K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 2009, 25, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Carlson, M. Yeast2.db: Affymetrix Yeast Genome 2.0 Array Annotation Data (Chip Yeast2), R package version 3.2.3: 2016. Available online: https://bioconductor.org/packages/release/data/annotation/html/yeast2.db.html (accessed on 10 November 2022).
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2011, 40, D700–D705. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling, R package version 1.16.0: 2022. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 10 November 2022).
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.-P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2020, 49, D325–D334. [Google Scholar] [CrossRef]
- Teixeira, M.C.; Viana, R.; Palma, M.; Oliveira, J.; Galocha, M.; Mota, M.N.; Couceiro, D.; Pereira, M.G.; Antunes, M.; Costa, I.V.; et al. YEASTRACT+: A portal for the exploitation of global transcription regulation and metabolic model data in yeast biotechnology and pathogenesis. Nucleic Acids Res 2022, 51, D785–D791. [Google Scholar] [CrossRef]
- Träff, K.L.; Jönsson, L.J.; Hahn-Hägerdal, B. Putative xylose and arabinose reductases in Saccharomyces cerevisiae. Yeast 2002, 19, 1233–1241. [Google Scholar] [CrossRef]
- Nelissen, B.; De Wachter, R.; Goffeau, A. Classification of all putative permeases and other membrane plurispanners of the major facilitator superfamily encoded by the complete genome of Saccharomyces cerevisiae. FEMS Microbiol. Rev. 1997, 21, 113–134. [Google Scholar] [CrossRef]
- Sousa, M.; Duarte, A.M.; Fernandes, T.R.; Chaves, S.R.; Pacheco, A.; Leao, C.; Corte-Real, M.; Sousa, M.J. Genome-wide identification of genes involved in the positive and negative regulation of acetic acid-induced programmed cell death in Saccharomyces cerevisiae. BMC Genom. 2013, 14, 838. [Google Scholar] [CrossRef] [Green Version]
- Sardi, M.; Rovinskiy, N.; Zhang, Y.; Gasch, A.P. Leveraging genetic-background effects in Saccharomyces cerevisiae to improve lignocellulosic hydrolysate tolerance. Appl Env. Microbiol 2016, 82, 5838–5849. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, I.M.; Zanotto-Filho, A.; Moreira, J.C.; Bonatto, D.; Henriques, J.A. The role of two putative nitroreductases, Frm2p and Hbn1p, in the oxidative stress response in Saccharomyces cerevisiae. Yeast 2010, 27, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hacisalihoglu, B.; Holyavkin, C.; Topaloglu, A.; Kisakesen, H.I.; Cakar, Z.P. Genomic and transcriptomic analysis of a coniferyl aldehyde-resistant Saccharomyces cerevisiae strain obtained by evolutionary engineering. FEMS Yeast Res. 2019, 19, foz021. [Google Scholar] [CrossRef] [PubMed]
- Ask, M.; Bettiga, M.; Mapelli, V.; Olsson, L. The influence of HMF and furfural on redox-balance and energy-state of xylose-utilizing Saccharomyces cerevisiae. Biotechnol. Biofuels. 2013, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Niño, M.; Pulido, S.; Stefanoska, D.; Pérez, C.; González-Ramos, D.; van Maris, A.J.A.; Marchal, K.; Nevoigt, E.; Swinnen, S. Identification of novel genes involved in acetic acid tolerance of Saccharomyces cerevisiae using pooled-segregant RNA sequencing. FEMS Yeast Res. 2018, 18, foy100. [Google Scholar] [CrossRef] [PubMed]
- Klinke, H.B.; Olsson, L.; Thomsen, A.B.; Ahring, B.K. Potential inhibitors from wet oxidation of wheat straw and their effect on ethanol production of Saccharomyces cerevisiae: Wet oxidation and fermentation by yeast. Biotechnol Bioeng 2003, 81, 738–747. [Google Scholar] [CrossRef]
- Larsson, S. Ethanol from Lignocellulose—Fermentation Inhibitors, Detoxification and Genetic Engineering of Saccharomyces cerevisiae for Enhanced Resistance. Ph.D. Thesis, Lund University, Lund, Sweden, 2000. [Google Scholar]
- Larroy, C.; Fernández, M.R.; González, E.; Parés, X.; Biosca, J.A. Characterization of the Saccharomyces cerevisiae YMR318C (ADH6) gene product as a broad specificity NADPH-dependent alcohol dehydrogenase: Relevance in aldehyde reduction. Biochem. J. 2002, 361, 163–172. [Google Scholar] [CrossRef]
- Liu, Z.L.; Moon, J.; Andersh, B.J.; Slininger, P.J.; Weber, S. Multiple gene-mediated NAD(P)H-dependent aldehyde reduction is a mechanism of in situ detoxification of furfural and 5-hydroxymethylfurfural by Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2008, 81, 743–753. [Google Scholar] [CrossRef]
- Li, X.; Yang, R.H.; Ma, M.G.; Wang, X.; Tang, J.; Zhao, X.X.; Zhang, X.P. A novel aldehyde reductase encoded by YML131W from Saccharomyces cerevisiae confers tolerance to furfural derived from lignocellulosic biomass conversion. Bioenerg. Res. 2015, 8, 119–129. [Google Scholar] [CrossRef]
- Ask, M.; Bettiga, M.; Duraiswamy, V.R.; Olsson, L. Pulsed addition of HMF and furfural to batch-grown xylose-utilizing Saccharomyces cerevisiae results in different physiological responses in glucose and xylose consumption phase. Biotechnol. Biofuels. 2013, 6, 181. [Google Scholar] [CrossRef] [Green Version]
- Massey, V.; Schopfer, L.M. Reactivity of old yellow enzyme with alpha-NADPH and other pyridine nucleotide derivatives. J. Biol. Chem. 1986, 261, 1215–1222. [Google Scholar] [CrossRef]
- Alriksson, B.; Horváth, I.S.; Jönsson, L.J. Overexpression of Saccharomyces cerevisiae transcription factor and multidrug resistance genes conveys enhanced resistance to lignocellulose-derived fermentation inhibitors. Process Biochem. 2010, 45, 264–271. [Google Scholar] [CrossRef]
- Piper, P.; Mahe, Y.; Thompson, S.; Pandjaitan, R.; Holyoak, C.; Egner, R.; Muhlbauer, M.; Coote, P.; Kuchler, K. The pdr12 ABC transporter is required for the development of weak organic acid resistance in yeast. EMBO J. 1998, 17, 4257–4265. [Google Scholar] [CrossRef] [Green Version]
- Sasano, Y.; Watanabe, D.; Ukibe, K.; Inai, T.; Ohtsu, I.; Shimoi, H.; Takagi, H. Overexpression of the yeast transcription activator Msn2 confers furfural resistance and increases the initial fermentation rate in ethanol production. J. Biosci. Bioeng. 2012, 113, 451–455. [Google Scholar] [CrossRef]
- Mertens, J.A.; Skory, C.D.; Nichols, N.N.; Hector, R.E. Impact of stress-response related transcription factor overexpression on lignocellulosic inhibitor tolerance of Saccharomyces cerevisiae environmental isolates. Biotechnol. Prog. 2021, 37, e3094. [Google Scholar] [CrossRef]
- Herrero, E.; Ros, J.; Belli, G.; Cabiscol, E. Redox control and oxidative stress in yeast cells. Biochim. Biophys. Acta 2008, 1780, 1217–1235. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. Gene-Specific Translational Control of the Yeast Gcn4 Gene by Phosphorylation of Eukaryotic Initiation Factor-Ii. Mol. Microbiol. 1993, 10, 215–223. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 2005, 59, 407–450. [Google Scholar] [CrossRef]
- Natarajan, K.; Meyer, M.R.; Jackson, B.M.; Slade, D.; Roberts, C.; Hinnebusch, A.G.; Marton, M.J. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol. Cell Biol. 2001, 21, 4347–4368. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Liu, Z.L. Comparative transcriptome profiling analyses during the lag phase uncover YAP1, PDR1, PDR3, RPN4, and HSF1 as key regulatory genes in genomic adaptation to the lignocellulose derived inhibitor HMF for Saccharomyces cerevisiae. BMC Genom. 2010, 11, 660. [Google Scholar] [CrossRef] [Green Version]
- Dohlman, H.G.; Thorner, J.W. Regulation of G protein-initiated signal transduction in yeast: Paradigms and principles. Annu. Rev. Biochem. 2001, 70, 703–754. [Google Scholar] [CrossRef]
- Shenhar, G.; Kassir, Y. A positive regulator of mitosis, Sok2, functions as a negative regulator of meiosis in Saccharomyces cerevisiae. Mol. Cell Biol. 2001, 21, 1603–1612. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SSF Experiments | Fed-Batch Cultivation | |||
|---|---|---|---|---|
| Solids (% of WIS) | Liquid (g/L) | Liquid (g/L) | ||
| Glucan | 54.6 | Glucose | 19.8 | 35.3 |
| Mannan | - | Mannose | 33.9 | 20.4 |
| Galactan | - | Galactose | 5.9 | 4.5 |
| Xylan | - | Xylose | 12.4 | 8.1 |
| Lignin | 39.3 | HMF | 1.9 | 3.7 |
| Furfural | 1.2 | 3.0 | ||
| Acetic acid | 4.3 | 5.1 | ||
| Yeast Cultivated in | Ethanol (g/L) | Ethanol Yield (% of Theoretical) | Initial Ethanol Prod. 1 (g/L.h) | Acetate (g/L) | Xylitol (g/L) | Glycerol (g/L) |
|---|---|---|---|---|---|---|
| SH | 40.9 ± 0.1 | 84.8 ± 0.3 | 1.98 ± 0.67 | 3.8 ± 0.8 | 2.4 ± 2.1 | 3.6 ± 0.4 |
| SM | 39.3 ± 0.3 | 81.6 ± 0.6 | 0.60 ± 0.06 | 3.5 ± 0.0 | 1.3 ± 2.9 | 2.2 ± 0.4 |
| GO Term (Biological Process) | Fold Enrichment of GO Term in the 9 h Dataset Compared to the Full S. cerevisiae Dataset | FDR (False Discovery Rate) | Genes in 9 h Set with the GO Term |
|---|---|---|---|
| Galactose catabolic process via UDP-galactose (GO:0033499) | >100 | 4.42 × 10−3 | GAL1; GAL7; GAL10 |
| Galactose catabolic process (GO:0019388) | 98.64 | 7.76 × 10−3 | GAL1; GAL7; GAL10 |
| Galactose metabolic process (GO:0006012) | 80.94 | 9.10 × 10−4 | GAL1; GAL2; GAL7; GAL10 |
| Hexose catabolic process (GO:0019320) | 65.76 | 1.51 × 10−2 | GAL1; GAL7; GAL10 |
| Monosaccharide catabolic process (GO:0046365) | 43.84 | 2.77 × 10−2 | GAL1; GAL7; GAL10 |
| Hexose metabolic process (GO:0019318) | 20.23 | 2.55 × 10−2 | GAL1; GAL2; GAL7; GAL10 |
| Monosaccharide metabolic process (GO:0005996) | 16.97 | 4.06 × 10−2 | GAL1; GAL2; GAL7; GAL10 |
| Inorganic ion transmembrane transport (GO:0098660) | 10.64 | 3.71 × 10−3 | ARN1; ATR1; CTR1; GAL2; HXT2; SUL1; ZRT1 |
| Ion transmembrane transport (GO:0034220) | 7.37 | 1.72 × 10−2 | ARN1; ATR1; AQY1; CTR1; GAL2; HXT2; PTR1; SUL1; ZRT1 |
| Transmembrane transport (GO:0055085) | 6.40 | 1.32 × 10−3 | ARN1; ATR1; AQY1; CTR1; FLR1; GAL2; HXT2; PTR2; SUL1; YHK8; ZRT1; |
| Ion transport (GO:0006811) | 6.17 | 1.59 × 10−2 | ARN1; ATR1; CTR1; GAL2; HXT2; SUL1; YHK8; ZRT1; |
| Nitrogen compound metabolic process (GO:0006807) | 0.09 | 8.55 × 10−3 | RCK1 |
| Gene | Fold Change (log2FC) | GO Category/Function | Previous Knowledge on Inhibitor/stress Tolerance |
|---|---|---|---|
| YHK8 (YHR048W) | 2.94 (9 h) 2.21 (17 h) | Member of the multidrug permease homolog family [42] Believed to be involved in xenobiotic detoxification by transmembrane export; putative plasma membrane localization | Deletion reported to increase tolerance to acetate [43] Deletion reported to increase sensitivity to wheat straw hydrolysate [20] Overexpression shown to reduce the lag phase in wheat straw hydrolysate [20] |
| OYE3 | 2.29 (9 h) 2.39 (17 h) | NADPH dehydrogenase with a role in apoptosis | Deletion reported to increase tolerance to acetate [43] Overexpression reported to decrease fitness in hydrolysate toxin cocktails [44] |
| HBN1 | 1.87 (9 h) 3.03 (17 h) | Putative oxidoreductase acting on NAD(P)H and nitrogenous group as acceptor Hbn1p nitroreductases also influences the response to oxidative stress in S. cerevisiae yeast by modulating the GSH contents and antioxidant enzymatic activities [45] | Upregulated genes in a coniferyl aldehyde-resistant strain [46] as well as in the presence of HMF and furfural [47] Upregulated during short-term adaptation on wheat straw hydrolysate [20] An HBN1 locus with a synonymous sequence variant was found to be significantly differentially expressed between a yeast strain with superior acetate tolerance and a strain with inferior tolerance [48] |
| YPR159C-A | 2.78 (9 h) 3.69 (17 h) | Unknown | Deletion increases tolerance to acetate [43] Overexpression decreases fitness in hydrolysate toxin cocktails [44] |
| CTR1 | −3.13 (9 h) −4.59 (17 h) | Copper ion transmembrane transporter that has role in copper ion import | Overexpression decreases fitness in hydrolysate toxin cocktails [44] |
| RCK1 | −2.91 (9 h) −3.23 (17 h) | Kinase involved in regulation of meiotic nuclear division | Deletion increases tolerance to acetate [43] |
| Transcription Factor | Number of Up-/Downregulated Genes with Documented Regulation by the Given TF | |
|---|---|---|
| 9 h | 17 h | |
| Msn2p, stress-responsive transcriptional activator | 11/21 1 | 86/297 |
| Ste12p, TF factor that is activated by a MAPK signaling cascade | 10/21 | 71/297 |
| Yap1p, TF required for oxidative stress tolerance | 8/21 | 39/297 |
| Sok2p, Nuclear protein that negatively regulates pseudohyphal differentiation | 7/21 | 48/297 |
| Msn4p, stress-responsive transcriptional activator | 7/21 | 31/297 |
| Gcn4p, bZIP transcriptional activator of amino acid biosynthetic genes | 6/21 | 82/297 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, J.R.M.; Wiman, M.; Heer, D.; Brink, D.P.; Sauer, U.; Hahn-Hägerdal, B.; Lidén, G.; Gorwa-Grauslund, M.F. Physiological and Molecular Characterization of Yeast Cultures Pre-Adapted for Fermentation of Lignocellulosic Hydrolysate. Fermentation 2023, 9, 72. https://doi.org/10.3390/fermentation9010072
Almeida JRM, Wiman M, Heer D, Brink DP, Sauer U, Hahn-Hägerdal B, Lidén G, Gorwa-Grauslund MF. Physiological and Molecular Characterization of Yeast Cultures Pre-Adapted for Fermentation of Lignocellulosic Hydrolysate. Fermentation. 2023; 9(1):72. https://doi.org/10.3390/fermentation9010072
Chicago/Turabian StyleAlmeida, João R. M., Magnus Wiman, Dominik Heer, Daniel P. Brink, Uwe Sauer, Bärbel Hahn-Hägerdal, Gunnar Lidén, and Marie F. Gorwa-Grauslund. 2023. "Physiological and Molecular Characterization of Yeast Cultures Pre-Adapted for Fermentation of Lignocellulosic Hydrolysate" Fermentation 9, no. 1: 72. https://doi.org/10.3390/fermentation9010072
APA StyleAlmeida, J. R. M., Wiman, M., Heer, D., Brink, D. P., Sauer, U., Hahn-Hägerdal, B., Lidén, G., & Gorwa-Grauslund, M. F. (2023). Physiological and Molecular Characterization of Yeast Cultures Pre-Adapted for Fermentation of Lignocellulosic Hydrolysate. Fermentation, 9(1), 72. https://doi.org/10.3390/fermentation9010072