Interfacial Spin Manipulation of Nickel-Quinonoid Complex Adsorbed on Co(001) Substrate


Abstract
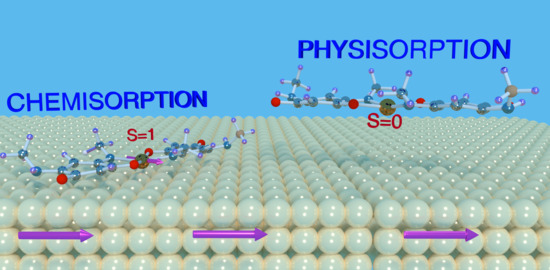
:
1. Introduction
2. Computational Methodology
3. Results and Discussion
3.1. Optimized Geometry
3.2. Electronic Structure and Magnetic Properties
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| DFT | Density Functional Theory |
| XMCD | X-ray magnetic circular dichroism |
| SP-STM | Spin-polarized scanning tunneling microscopy |
| TM | Transition metal |
| TM-P | transition-metal porphyrin |
| TM-Pc | Transition-metal phthalocyanine |
| NiQ | Ni(II)-quinonoid |
| MeOH | Methanol |
| VASP | Vienna Ab-initio Simulation Package |
| PAW | Projector augmented plane wave |
| GGA | Generalized gradient approximation |
| LF | Ligand field |
| DOS | Density of states |
| PDOS | Partial density of states |
Appendix A. Physisorption of NiQ on Co(001)


References
- Bogani, L.; Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 2008, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Moodera, J.S.; Koopmans, B.; Oppeneer, P.M. On the path toward organic spintronics. MRS Bull. 2014, 39, 578–581. [Google Scholar] [CrossRef] [Green Version]
- Gütlich, P.; Goodwin, H.A. Spin Crossover in Transition Metal Compounds I; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Hao, H.; Zheng, X.; Song, L.; Wang, R.; Zeng, Z. Electrostatic spin crossover in a molecular junction of a single-molecule magnet Fe2. Phys. Rev. Lett. 2012, 108, 017202. [Google Scholar] [CrossRef] [PubMed]
- Coronado, E.; Palacio, F.; Veciana, J. Molecule-Based Magnetic Materials. Angew. Chem. Int. Ed. 2003, 42, 2570–2572. [Google Scholar] [CrossRef] [PubMed]
- Gatteschi, D.; Sessoli, R.; Villain, J. Molecular Nanomagnets; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Benelli, C.; Gatteschi, D. Introduction to Molecular Magnetism—From Transition Metals to Lanthanides; Wiley–VCH: Weinheim, Germany, 2015. [Google Scholar]
- Banerjee, H.; Chakraborty, S.; Saha-Dasgupta, T. Design and Control of Cooperativity in Spin-Crossover in Metal-Organic Complexes: A Theoretical Overview. Inorganics 2017, 5, 47. [Google Scholar] [CrossRef]
- Kumar, K.S.; Ruben, M. Emerging trends in spin crossover (SCO) based functional materials and devices. Coord. Chem. Rev. 2017, 346, 176–205. [Google Scholar] [CrossRef]
- Molnár, G.; Rat, S.; Salmon, L.; Nicolazzi, W.; Bousseksou, A. Spin crossover nanomaterials: From fundamental concepts to devices. Adv. Mater. 2018, 30, 1703862. [Google Scholar] [CrossRef] [PubMed]
- Auwärter, W.; Écija, D.; Klappenberger, F.; Barth, J.V. Porphyrins at interfaces. Nat. Chem. 2015, 7, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, J.M. Surface chemistry of porphyrins and phthalocyanines. Surf. Sci. Rep. 2015, 70, 259–379. [Google Scholar] [CrossRef]
- Scheybal, A.; Ramsvik, T.; Bertschinger, R.; Putero, M.; Nolting, F.; Jung, T.A. Induced magnetic ordering in a molecular monolayer. Chem. Phys. Lett. 2005, 411, 214–220. [Google Scholar] [CrossRef]
- Gambardella, P.; Stepanow, S.; Dmitriev, A.; Honolka, J.; de Groot, F.M.F.; Lingenfelder, M.; Gupta, S.S.; Sarma, D.D.; Bencok, P.; Stanescu, S.; et al. Supramolecular control of the magnetic anisotropy in two-dimensional high-spin Fe arrays at a metal interface. Nat. Mater. 2009, 8, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.S.; Gatteschi, D. Molecule-based magnets. Chem. Soc. Rev. 2011, 40, 3065–3066. [Google Scholar] [CrossRef]
- Raman, K.V.; Kamerbeek, A.M.; Mukherjee, A.; Atodiresei, N.; Sen, T.K.; Lazić, P.; Caciuc, V.; Michel, R.; Stalke, D.; Mandal, S.K.; et al. Interface-engineered templates for molecular spin memory devices. Nature 2013, 493, 509. [Google Scholar] [CrossRef] [PubMed]
- Djeghloul, F.; Ibrahim, F.; Cantoni, M.; Bowen, M.; Joly, L.; Boukari, S.; Ohresser, P.; Bertran, F.; Le Févre, P.; Thakur, P. Direct observation of a highly spin-polarized organic spinterface at room temperature. Sci. Rep. 2013, 3, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steil, S.; Großmann, N.; Laux, M.; Ruffing, A.; Steil, D.; Wiesenmayer, M.; Mathias, S.; Monti, O.L.A.; Cinchetti, M.; Aeschlimann, M. Spin-dependent trapping of electrons at spinterfaces. Nat. Phys. 2013, 9, 242–247. [Google Scholar] [CrossRef]
- Bairagi, K.; Bellec, A.; Repain, V.; Chacon, C.; Girard, Y.; Garreau, Y.; Lagoute, J.; Rousset, S.; Breitwieser, R.; Hu, Y.C. Tuning the magnetic anisotropy at a molecule-metal interface. Phys. Rev. Lett. 2015, 114, 247203. [Google Scholar] [CrossRef] [PubMed]
- Barraud, C.; Bouzehouane, K.; Deranlot, C.; Fusil, S.; Jabbar, H.; Arabski, J.; Rakshit, R.; Kim, D.J.; Kieber, C.; Boukari, S. Unidirectional spin-dependent molecule-ferromagnet hybridized states anisotropy in cobalt phthalocyanine based magnetic tunnel junctions. Phys. Rev. Lett. 2015, 114, 206603. [Google Scholar] [CrossRef] [PubMed]
- Sanvito, S. Molecular spintronics. Chem. Soc. Rev. 2011, 40, 3336–3355. [Google Scholar] [CrossRef]
- Rocha, A.R.; Garcia-Suarez, V.M.; Bailey, S.W.; Lambert, C.J.; Ferrer, J.; Sanvito, S. Towards molecular spintronics. Nat. Mater. 2005, 4, 335. [Google Scholar] [CrossRef]
- Wende, H.; Bernien, M.; Luo, J.; Sorg, C.; Ponpandian, N.; Kurde, J.; Miguel, J.; Piantek, M.; Xu, X.; Eckhold, P.; et al. Substrate-induced magnetic ordering and switching of iron porphyrin molecules. Nat. Mater. 2007, 6, 516. [Google Scholar] [CrossRef]
- Lach, S.; Altenhof, A.; Tarafder, K.; Schmitt, F.; Ali, M.E.; Vogel, M.; Sauther, J.; Oppeneer, P.M.; Ziegler, C. Metal-organic hybrid interface states of a ferromagnet/organic semiconductor hybrid junction as basis for engineering spin injection in organic spintronics. Adv. Funct. Mater. 2012, 22, 989–997. [Google Scholar] [CrossRef]
- Wäckerlin, C.; Tarafder, K.; Siewert, D.; Girovsky, J.; Hählen, T.; Iacovita, C.; Kleibert, A.; Nolting, F.; Jung, T.A.; Oppeneer, P.M.; et al. On-surface coordination chemistry of planar molecular spin systems: Novel magnetochemical effects induced by axial ligands. Chem. Sci. 2012, 3, 3154–3160. [Google Scholar] [CrossRef]
- Wäckerlin, C.; Tarafder, K.; Girovsky, J.; Nowakowski, J.; Hählen, T.; Shchyrba, A.; Siewert, D.; Kleibert, A.; Nolting, F.; Oppeneer, P.M.; et al. Ammonia Coordination Introducing a Magnetic Moment in an On-Surface Low-Spin Porphyrin. Angew. Chem. Int. Ed. 2013, 125, 4666–4669. [Google Scholar] [CrossRef] [Green Version]
- Cornia, A.; Mannini, M.; Sainctavit, P.; Sessoli, R. Chemical strategies and characterization tools for the organization of single molecule magnets on surfaces. Chem. Soc. Rev. 2011, 40, 3076–3091. [Google Scholar] [CrossRef] [PubMed]
- Iacovita, C.; Rastei, M.V.; Heinrich, B.W.; Brumme, T.; Kortus, J.; Limot, L.; Bucher, J.P. Visualizing the spin of individual cobalt-phthalocyanine molecules. Phys. Rev. Lett. 2008, 101, 116602. [Google Scholar] [CrossRef]
- Hermanns, C.F.; Tarafder, K.; Bernien, M.; Krüger, A.; Chang, Y.M.; Oppeneer, P.M.; Kuch, W. Magnetic coupling of porphyrin molecules through graphene. Adv. Mater. 2013, 25, 3473–3477. [Google Scholar] [CrossRef] [PubMed]
- Lodi Rizzini, A.; Krull, C.; Balashov, T.; Mugarza, A.; Nistor, C.; Yakhou, F.; Sessi, V.; Klyatskaya, S.; Ruben, M.; Stepanow, S.; et al. Exchange Biasing Single Molecule Magnets: Coupling of TbPc2 to Antiferromagnetic Layers. Nano Lett. 2012, 12, 5703–5707. [Google Scholar] [CrossRef] [PubMed]
- Girovsky, J.; Nowakowski, J.; Ali, M.E.; Baljozovic, M.; Rossmann, H.R.; Nijs, T.; Aeby, E.A.; Nowakowska, S.; Siewert, D.; Srivastava, G.; et al. Long-range ferrimagnetic order in a two-dimensional supramolecular Kondo lattice. Nat. Commun. 2017, 8, 15388. [Google Scholar] [CrossRef] [Green Version]
- Gruber, M.; Berndt, R. Manipulation of Cyclohexene-Based Organic Molecules on Various Metallic Substrates. J. Phys. Chem. C 2016, 120, 18642–18650. [Google Scholar] [CrossRef]
- Flechtner, K.; Kretschmann, A.; Steinrück, H.P.; Gottfried, J.M. NO-Induced Reversible Switching of the Electronic Interaction between a Porphyrin-Coordinated Cobalt Ion and a Silver Surface. J. Am. Chem. Soc. 2007, 129, 12110–12111. [Google Scholar] [CrossRef]
- Bernien, M.; Miguel, J.; Weis, C.; Ali, M.E.; Kurde, J.; Krumme, B.; Panchmatia, P.M.; Sanyal, B.; Piantek, M.; Srivastava, P.; et al. Tailoring the nature of magnetic coupling of Fe-porphyrin molecules to ferromagnetic substrates. Phys. Rev. Lett. 2009, 102, 047202. [Google Scholar] [CrossRef] [PubMed]
- Ballav, N.; Wäckerlin, C.; Siewert, D.; Oppeneer, P.M.; Jung, T.A. Emergence of On-Surface Magnetochemistry. J. Phys. Chem. Lett. 2013, 4, 2303–2311. [Google Scholar] [CrossRef] [Green Version]
- Herper, H.C.; Bernien, M.; Bhandary, S.; Hermanns, C.F.; Krüger, A.; Miguel, J.; Weis, C.; Schmitz-Antoniak, C.; Krumme, B.; Bovenschen, D.; et al. Iron porphyrin molecules on Cu (001): Influence of adlayers and ligands on the magnetic properties. Phys. Rev. B 2013, 87, 174425. [Google Scholar] [CrossRef]
- Bhandary, S.; Brena, B.; Panchmatia, P.M.; Brumboiu, I.; Bernien, M.; Weis, C.; Krumme, B.; Etz, C.; Kuch, W.; Wende, H.; et al. Manipulation of spin state of iron porphyrin by chemisorption on magnetic substrates. Phys. Rev. B 2013, 88, 024401. [Google Scholar] [CrossRef] [Green Version]
- Kar, P.; Yoshida, M.; Shigeta, Y.; Usui, A.; Kobayashi, A.; Minamidate, T.; Matsunaga, N.; Kato, M. Methanol-Triggered Vapochromism Coupled with Solid-State Spin Switching in a Nickel (II)-Quinonoid Complex. Angew. Chem. Int. Ed. 2017, 129, 2385–2389. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Ali, M.E.; Sanyal, B.; Oppeneer, P.M. Tuning the magnetic interaction between manganese porphyrins and ferromagnetic Co substrate through dedicated control of the adsorption. J. Phys. Chem. C 2009, 113, 14381–14383. [Google Scholar] [CrossRef]
- Tarafder, K.; Kanungo, S.; Oppeneer, P.M.; Saha-Dasgupta, T. Pressure and temperature control of spin-switchable metal-organic coordination polymers from ab initio calculations. Phys. Rev. Lett. 2012, 109, 077203. [Google Scholar] [CrossRef]
- Ali, M.E.; Sanyal, B.; Oppeneer, P.M. Electronic Structure, Spin-States, and Spin-Crossover Reaction of Heme-Related Fe-Porphyrins: A Theoretical Perspective. J. Phys. Chem. B 2012, 116, 5849–5859. [Google Scholar] [CrossRef] [PubMed]
- Lebègue, S.; Pillet, S.; Ángyán, J.G. Modeling spin-crossover compounds by periodic DFT+U approach. Phys. Rev. B 2008, 78, 024433. [Google Scholar] [CrossRef]
- Panchmatia, P.M.; Ali, M.E.; Sanyal, B.; Oppeneer, P.M. Halide Ligated Iron Porphines: A DFT+U and UB3LYP Study. J. Phys. Chem. A 2010, 114, 13381–13387. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, P.; Kanungo, S.; Saha-Dasgupta, T.; Oppeneer, P.M. Two-step spin-switchable tetranuclear Fe(II) molecular solid: Ab initio theory and predictions. Phys. Rev. B 2013, 88, 020408. [Google Scholar] [CrossRef]
- Dion, M.; Rydberg, H.; Schröder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals Density Functional for General Geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gas phase | HOLLOW | TOP | |
|---|---|---|---|
| Ni–O1 | 1.8562 | 2.1151 | 2.1156 |
| Ni–O2 | 1.8558 | 2.1158 | 2.1166 |
| Ni–N1 | 1.8782 | 2.0290 | 1.8778 |
| Ni–N2 | 1.8799 | 2.0295 | 1.8778 |
| Average | 1.8683 | 2.0724 | 1.9967 |
| Magnetic Ni moment | 0.000 | 1.178 | 1.133 |
| Magnetic moment | 0.000 | 1.561 | 1.337 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, I.R.; Oppeneer, P.M.; Tarafder, K. Interfacial Spin Manipulation of Nickel-Quinonoid Complex Adsorbed on Co(001) Substrate. Magnetochemistry 2019, 5, 2. https://doi.org/10.3390/magnetochemistry5010002
Reddy IR, Oppeneer PM, Tarafder K. Interfacial Spin Manipulation of Nickel-Quinonoid Complex Adsorbed on Co(001) Substrate. Magnetochemistry. 2019; 5(1):2. https://doi.org/10.3390/magnetochemistry5010002
Chicago/Turabian StyleReddy, Indukuru Ramesh, Peter M. Oppeneer, and Kartick Tarafder. 2019. "Interfacial Spin Manipulation of Nickel-Quinonoid Complex Adsorbed on Co(001) Substrate" Magnetochemistry 5, no. 1: 2. https://doi.org/10.3390/magnetochemistry5010002
APA StyleReddy, I. R., Oppeneer, P. M., & Tarafder, K. (2019). Interfacial Spin Manipulation of Nickel-Quinonoid Complex Adsorbed on Co(001) Substrate. Magnetochemistry, 5(1), 2. https://doi.org/10.3390/magnetochemistry5010002