
EDTA and NTA Effectively Tune the Mineralization of Calcium Phosphate from Bulk Aqueous Solution


 and
and 
Abstract
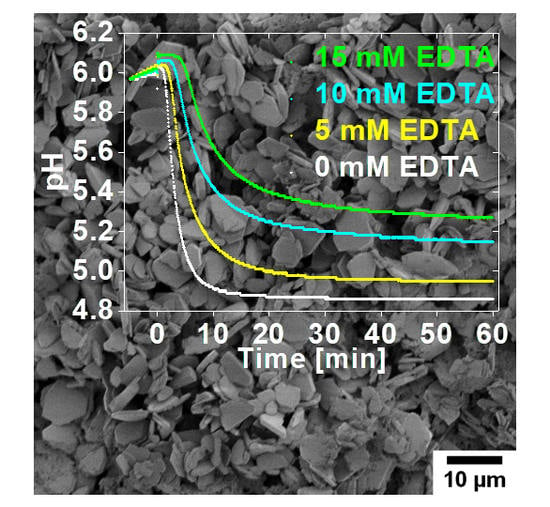
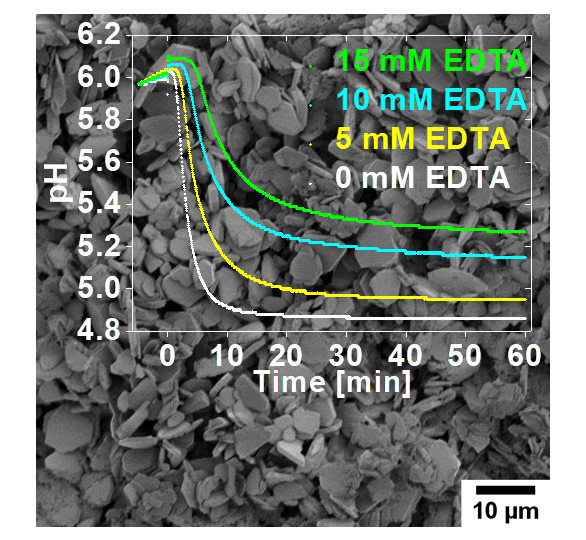
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.1.1. Nitrilotriacetic Acid Trisodium Salt Monohydrate
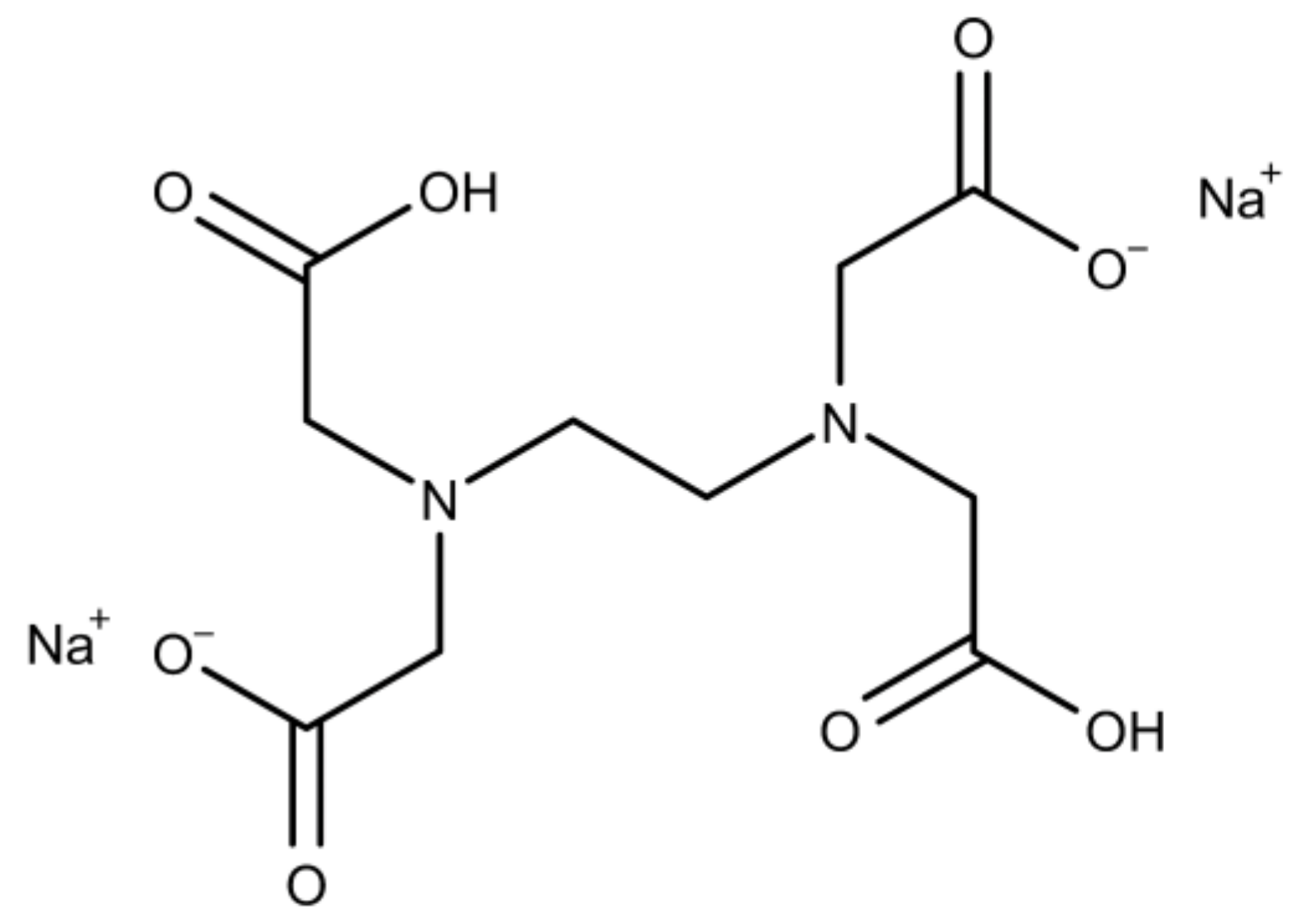
2.1.2. Ethylenediaminotetraacetic Acid Disodium Salt Dihydrate
2.2. Mineralization
2.3. Attenuated Total Reflectance–Fourier Transform Infrared Spectroscopy
2.4. Elemental Analysis
2.5. Nuclear Magnetic Resonance Spectroscopy
2.6. Scanning Electron Microscopy
2.7. X-ray Powder Diffraction
2.8. Conductivity Measurements
2.9. pH Measurements
3. Results
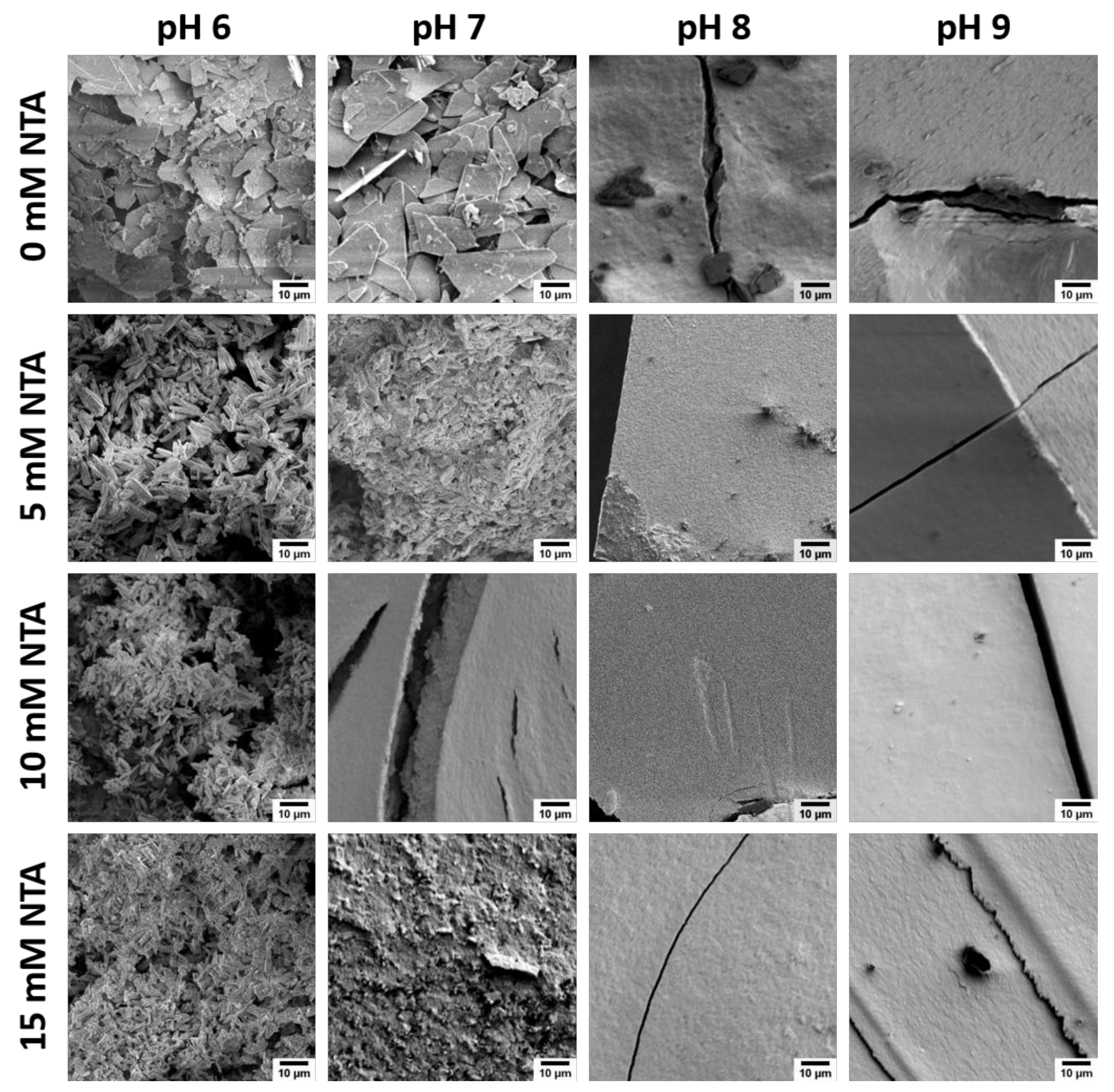
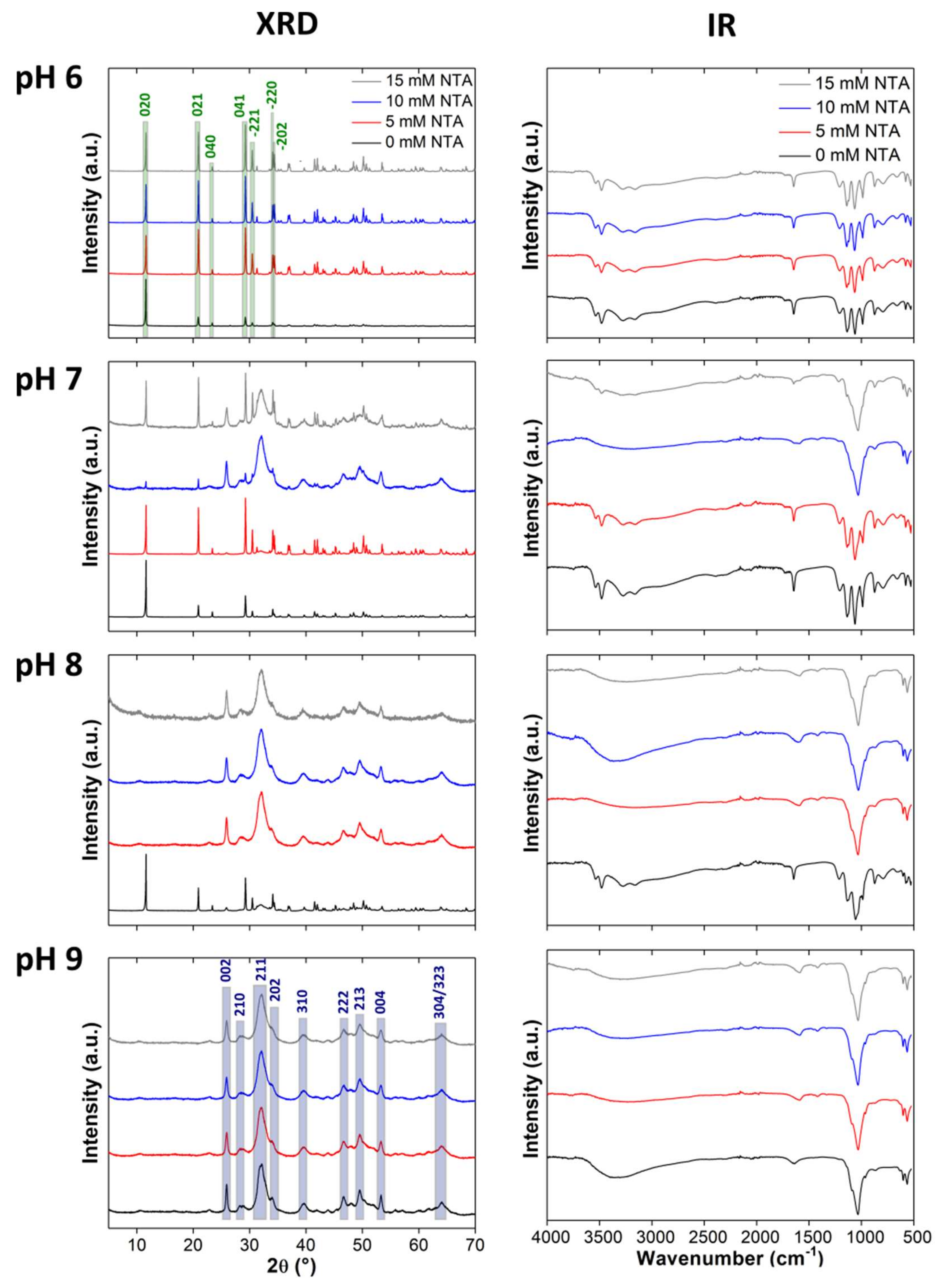
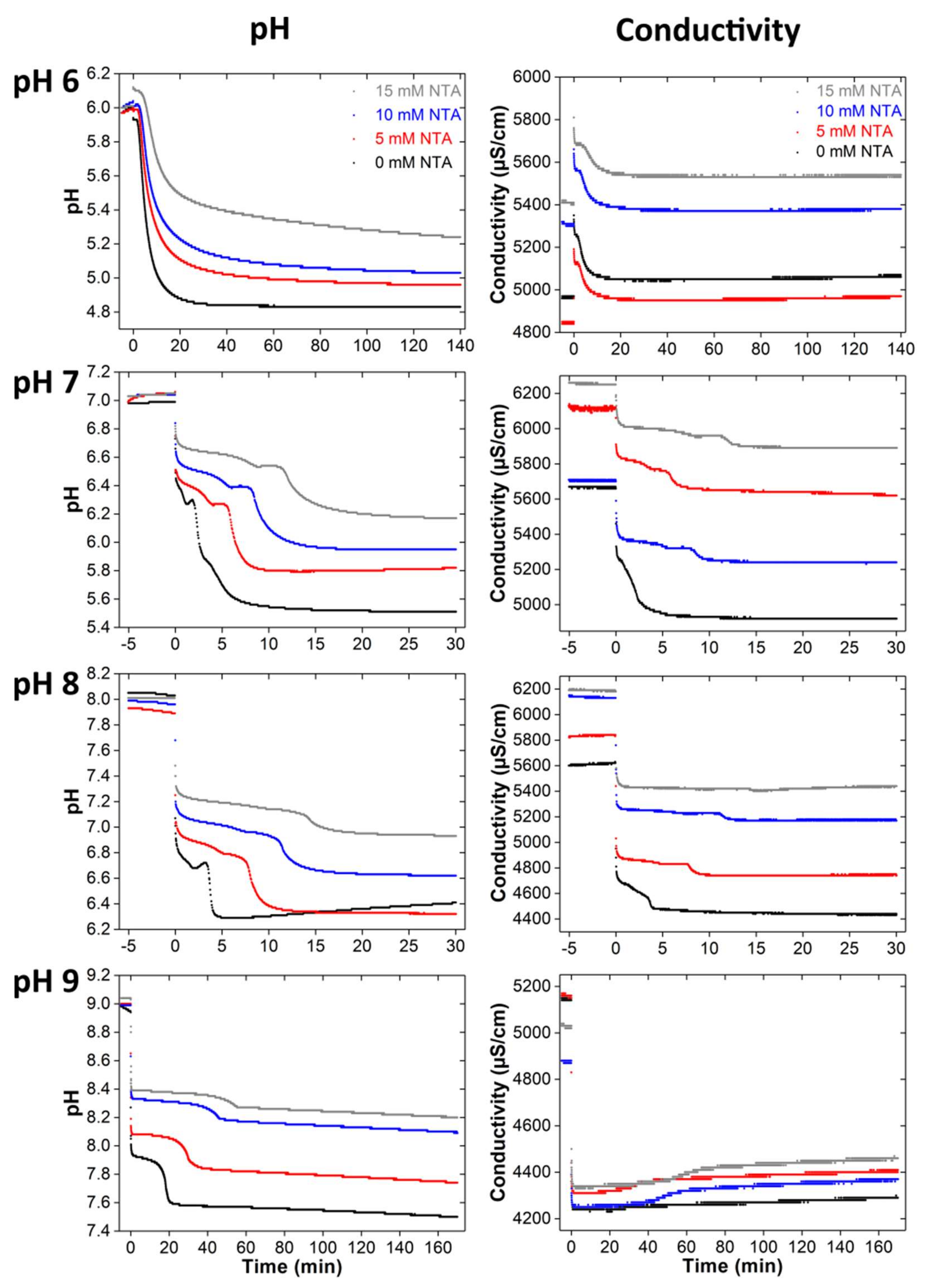
3.1. Calcium Phosphate Mineralization with NTA
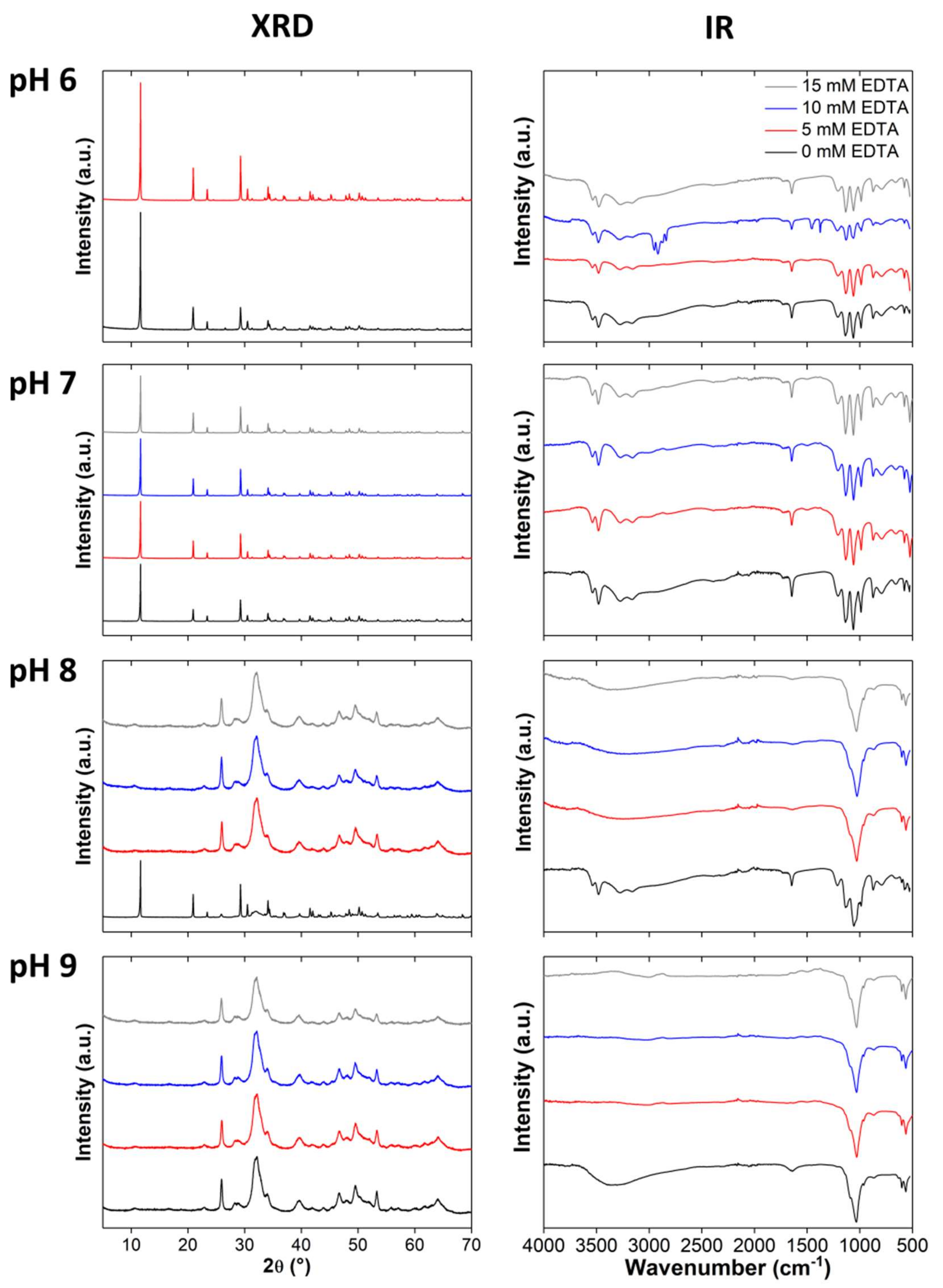
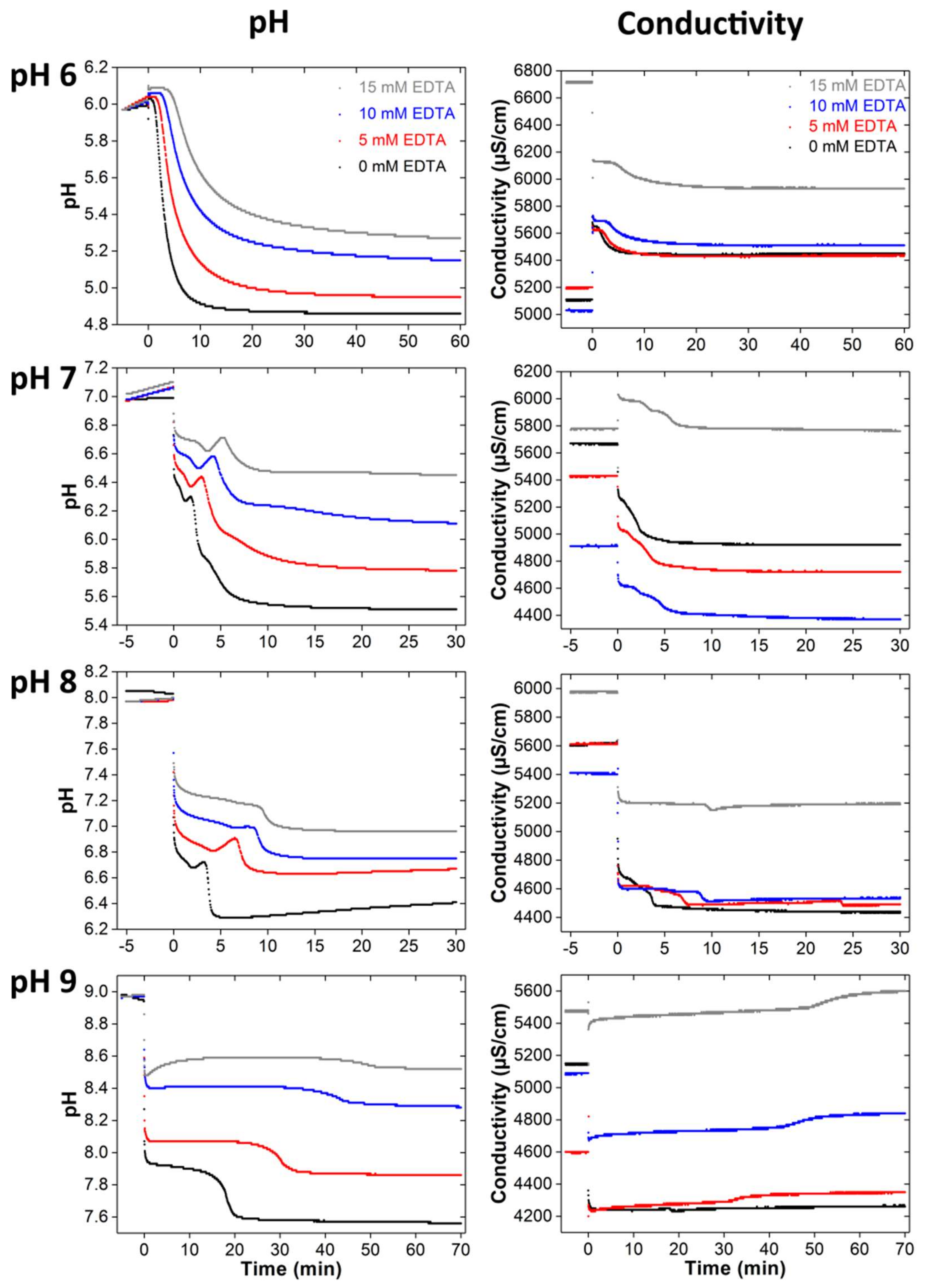
3.2. Clacium Phosphate Mineralization with EDTA
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

References
- Dey, A.; van den Hoogen, C.J.; Rosso, M.; Lousberg, N.; Hendrix, M.M.R.M.; Friedrich, H.; Ramírez-Rico, J.; Zuilhof, H.; de With, G.; Sommerdijk, N.A.J.M. Biomimetic mineralization of calcium phosphate on a functionalized porous silicon carbide biomaterial. ChemPlusChem 2012, 77, 694–699. [Google Scholar] [CrossRef]
- Du, C.; Cui, F.Z.; Zhang, W.; Feng, Q.L.; Zhu, X.D.; de Groot, K. Formation of calcium phosphate/collagen composites through mineralization of collagen matrix. J. Biomed. Mater. Res. Part A 2000, 50, 518–527. [Google Scholar] [CrossRef]
- Spoerke, E.D.; Stupp, S.I. Synthesis of a poly(l-lysine)-calcium phosphate hybrid on titanium surfaces for enhanced bioactivity. Biomaterials 2005, 26, 5120–5129. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, S.; Schuster, T.; Junginger, M.; Siekmeyer, G.; Taubert, A. Surface modification of nickel/titanium alloy and titanium surfaces via a polyelectrolyte multilayer/calcium phosphate hybrid coating. Macromol. Mater. Eng. 2010, 295, 535–543. [Google Scholar] [CrossRef]
- Nudelman, F.; Sommerdijk, N.A.J.M. Biomineralization as an inspiration for materials chemistry. Angew. Chem. Int. Ed. 2012, 51, 6582–6596. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V.; Epple, M. Biological and medical significance of calcium phosphates. Angew. Chem. Int. Ed. 2002, 41, 3130–3146. [Google Scholar] [CrossRef]
- Epple, M. Biomaterialien und Biomineralisation; Teubner: Wiesbaden, Germany, 2003. [Google Scholar]
- Schweizer, S.; Taubert, A. Polymer-controlled, bio-inspired calcium phosphate mineralization from aqueous solution. Macromol. Biosci. 2007, 7, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Bleek, K.; Taubert, A. New developments in polymer-controlled, bioinspired calcium phosphate mineralization from aqueous solution. Acta Biomater. 2013, 9, 6283–6321. [Google Scholar] [CrossRef] [PubMed]
- Bäuerlein, E.; Behrens, P.; Epple, M. Handbook of Biomineralization: Biomimetic and Bioinspired Chemistry; Wiley-VCH: Weinheim, Germany, 2007; ISBN 9783527318056. [Google Scholar]
- Bäuerlein, E.; Behrens, P.; Epple, M. Handbook of Biomineralization: Biological Aspects and Structure Formation; Wiley-VCH: Weinheim, Germany, 2007; ISBN 9783527318049. [Google Scholar]
- Bertoni, E.; Bigi, A.; Falini, G.; Panzavolta, S.; Roveri, N. Hydroxyapatite/polyacrylic acid nanocrystals. J. Mater. Chem. 1999, 9, 779–782. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Botter, R.; Panzavolta, S.; Rubini, K. α-Tricalcium phosphate hydrolysis to octacalcium phosphate: Effect of sodium polyacrylate. Biomaterials 2002, 23, 1849–1854. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Cojazzi, G.; Falini, G.; Panzavolta, S. Morphological and structural investigation of octacalcium phosphate hydrolysis in the presence of polyacrylic acids: Effect of relative molecular weights. Cryst. Growth Des. 2001, 1, 239–244. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Panzavolta, S.; Roveri, N. Biomimetic growth of hydroxyapatite on gelatin films doped with sodium polyacrylate. Biomacromolecules 2000, 1, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Boanini, E.; Borghi, M.; Cojazzi, G.; Panzavolta, S.; Roveri, N. Synthesis and hydrolysis of octacalcium phosphate: Effect of sodium polyacrylate. J. Inorg. Biochem. 1999, 75, 145–151. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Falini, G.; Panzavolta, S.; Roveri, N. Effect of sodium polyacrylate on the hydrolysis of octacalcium phosphate. J. Inorg. Biochem. 2000, 78, 227–233. [Google Scholar] [CrossRef]
- Bertoni, E.; Bigi, A.; Cojazzi, G.; Gandolfi, M.; Panzavolta, S.; Roveri, N. Nanocrystals of magnesium and fluoride substituted hydroxyapatite. J. Inorg. Biochem. 1998, 72, 29–35. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Gazzano, M.; Kojdecki, M.A.; Rubini, K. Microstructural investigation of hydroxyapatite—Polyelectrolyte composites. J. Mater. Chem. 2004, 14, 274–279. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Walsh, D.; Mann, S. Morphosynthesis of octacalcium phosphate hollow microspheres by polyelectrolyte-mediated crystallization. Angew. Chem. Int. Ed. 2002, 41, 2163–2166. [Google Scholar] [CrossRef]
- Meldrum, F.C.; Cölfen, H. Controlling mineral morphologies and structures in biological and synthetic systems. Chem. Rev. 2008, 108, 4332–4432. [Google Scholar] [CrossRef] [PubMed]
- Boanini, E.; Torricelli, P.; Gazzano, M.; Giardino, R.; Bigi, A. Nanocomposites of hydroxyapatite with aspartic acid and glutamic acid and their interaction with osteoblast-like cells. Biomaterials 2006, 27, 4428–4433. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, F.; Bomans, P.H.H.; George, A.; de With, G.; Sommerdijk, N.A.J.M. The role of the amorphous phase on the biomimetic mineralization of collagen. Faraday Discuss. 2012, 159, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Nancollas, G.H. Calcium orthophosphates: Crystallization and dissolution. Chem. Rev. 2008, 108, 4628–4669. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Morales, J.; Iafisco, M.; Delgado-López, J.M.; Sarda, S.; Drouet, C. Progress on the preparation of nanocrystalline apatites and surface characterization: Overview of fundamental and applied aspects. Prog. Cryst. Growth Charact. Mater. 2013, 59, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, A.S.; Beniash, E. Bioinspired synthesis of mineralized collagen fibrils. Cryst. Growth Des. 2008, 8, 3084–3090. [Google Scholar] [CrossRef] [PubMed]
- Olszta, M.J.; Cheng, X.; Jee, S.S.; Kumar, R.; Kim, Y.-Y.; Kaufman, M.J.; Douglas, E.P.; Gower, L.B. Bone structure and formation: A new perspective. Mater. Sci. Eng. R Rep. 2007, 58, 77–116. [Google Scholar] [CrossRef]
- Peytcheva, A.; Cölfen, H.; Schnablegger, H.; Antonietti, M. Calcium phosphate colloids with hierarchical structure controlled by polyaspartates. Colloid Polym. Sci. 2002, 280, 218–227. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Bracci, B.; Falini, G.; Rubini, K. Interaction of acidic poly-amino acids with octacalcium phosphate. J. Inorg. Biochem. 2003, 95, 291–296. [Google Scholar] [CrossRef]
- Bar-Yosef Ofir, P.; Govrin-Lippman, R.; Garti, N.; Füredi-Milhofer, H. The Influence of Polyelectrolytes on the formation and phase transformation of amorphous calcium phosphate. Cryst. Growth Des. 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Chen, J.; Burger, C.; Krishnan, C.V.; Chu, B.; Hsiao, B.S.; Glimcher, M.J. In vitro mineralization of collagen in demineralized fish bone. Macromol. Chem. Phys. 2005, 206, 43–51. [Google Scholar] [CrossRef]
- Ball, V.; Michel, M.; Boulmedais, F.; Hemmerle, J.; Haikel, Y.; Schaaf, P.; Voegel, J.C. Nucleation kinetics of calcium phosphates on polyelectrolyte multilayers displaying internal secondary structure. Cryst. Growth Des. 2005, 6, 327–334. [Google Scholar] [CrossRef]
- Krogstad, D.V.; Wang, D.; Lin-Gibson, S. Polyaspartic acid concentration controls the rate of calcium phosphate nanorod formation in high concentration systems. Biomacromolecules 2017, 18, 3106–3113. [Google Scholar] [CrossRef] [PubMed]
- Shkilnyy, A.; Gräf, R.; Hiebl, B.; Neffe, A.T.; Friedrich, A.; Hartmann, J.; Taubert, A. Unprecedented, low cytotoxicity of spongelike calcium phosphate/poly(ethylene imine) hydrogel composites. Macromol. Biosci. 2009, 9, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Shkilnyy, A.; Friedrich, A.; Tiersch, B.; Schöne, S.; Fechner, M.; Koetz, J.; Schläpfer, C.-W.; Taubert, A.; Schone, S.; Schlapfer, C.-W. Poly(ethylene imine)-controlled calcium phosphate mineralization. Langmuir 2008, 24, 2102–2109. [Google Scholar] [CrossRef] [PubMed]
- Shkilnyy, A.; Brandt, J.; Mantion, A.; Paris, O.; Schlaad, H.; Taubert, A. Calcium phosphate with a channel-like morphology by polymer templating. Chem. Mater. 2009, 21, 1572–1578. [Google Scholar] [CrossRef]
- Junginger, M.; Kita-Tokarczyk, K.; Schuster, T.; Reiche, J.; Schacher, F.; Müller, A.H.E.; Cölfen, H.; Taubert, A. Calcium phosphate mineralization beneath a polycationic monolayer at the air-water interface. Macromol. Biosci. 2010, 10, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Junginger, M.; Kübel, C.; Schacher, F.H.; Müller, A.H.E.; Taubert, A. Crystal structure and chemical composition of biomimetic calcium phosphate nanofibers. RSC Adv. 2013, 3, 11301–11308. [Google Scholar] [CrossRef]
- Junginger, M.; Bleek, K.; Kita-Tokarczyk, K.; Reiche, J.; Shkilnyy, A.; Schacher, F.; Müller, A.H.E.; Taubert, A. Calcium phosphate growth beneath a polycationic monolayer at the air–water interface: Effects of oscillating surface pressure on mineralization. Nanoscale 2010, 2, 2440–2446. [Google Scholar] [CrossRef] [PubMed]
- Ngankam, P.A.; Lavalle, P.; Szyk, L.; Decher, G.; Schaaf, P.; Cuisinier, F.J.G.; Cedex, S. Influence of polyelectrolyte multilayer films on calcium phosphate nucleation. J. Am. Chem. Soc. 2000, 122, 8998–9005. [Google Scholar] [CrossRef]
- Michel, M.; Arntz, Y.; Fleith, G.; Toquant, J.; Haikel, Y.; Voegel, J.C.; Schaaf, P.; Ball, V. Layer-by-layer self-assembled polyelectrolyte multilayers with embedded liposomes: Immobilized submicronic reactors for mineralization. Langmuir 2006, 22, 2358–2364. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gu, W.; Pan, H.; Jiang, S.; Tang, R. Stabilizing amorphous calcium phosphate phase by citrate adsorption. CrystEngComm 2014, 16, 1864–1867. [Google Scholar] [CrossRef]
- Martins, M.A.; Santos, C.; Almeida, M.M.; Costa, M.E.V. Hydroxyapatite micro- and nanoparticles: Nucleation and growth mechanisms in the presence of citrate species. J. Colloid Interface Sci. 2008, 318, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Iafisco, M.; Ramírez-Rodríguez, G.B.; Sakhno, Y.; Tampieri, A.; Martra, G.; Gómez-Morales, J.; Delgado-López, J.M. The growth mechanism of apatite nanocrystals assisted by citrate: Relevance to bone biomineralization. CrystEngComm 2015, 17, 507–511. [Google Scholar] [CrossRef]
- Hu, Y.Y.; Liu, X.P.; Ma, X.; Rawal, A.; Prozorov, T.; Akinc, M.; Mallapragada, S.K.; Schmidt-Rohr, K. Biomimetic self-assembling copolymer-hydroxyapatite nanocomposites with the nanocrystal size controlled by citrate. Chem. Mater. 2011, 23, 2481–2490. [Google Scholar] [CrossRef]
- Xie, B.; Nancollas, G.H. How to control the size and morphology of apatite nanocrystals in bone. Proc. Natl. Acad. Sci. USA 2010, 107, 22369–22370. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-Y.; Rawal, A.; Schmidt-Rohr, K. Strongly bound citrate stabilizes the apatite nanocrystals in bone. Proc. Natl. Acad. Sci. USA 2010, 107, 22425–22429. [Google Scholar] [CrossRef] [PubMed]
- Van Kemenade, M.J.J.M.; de Bruyn, P.L. A kinetic study of precipitation from supersaturated calcium phosphate solutions. J. Colloid Interface Sci. 1987, 118, 564–585. [Google Scholar] [CrossRef]
- Mekmene, O.; Quillard, S.; Rouillon, T.; Bouler, J.-M.; Piot, M.; Gaucheron, F. Effects of pH and Ca/P molar ratio on the quantity and crystalline structure of calcium phosphates obtained from aqueous solutions. Dairy Sci. Technol. 2009, 89, 301–316. [Google Scholar] [CrossRef]
- Margolis, H.C.; Kwak, S.; Yamazaki, H. Role of mineralization inhibitors in the regulation of hard tissue biomineralization: Relevance to initial enamel formation and maturation. Front. Physiol. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, Y.; Sato, T.; Okuwaki, A. Coating of hydroxyapatite on metal plates using thermal dissociation of calcium-EDTA chelate in phosphate solutions under hydrothermal conditions. J. Mater. Sci. Mater. Med. 1995, 6, 172–176. [Google Scholar] [CrossRef]
- Kandori, K.; Horigami, N.; Yasukawa, A.; Ishikawa, T. Texture and formation mechanism of fibrous calcium hydroxyapatite particles prepared by decomposition of calcium–EDTA chelates. J. Am. Ceram. Soc. 1997, 80, 1157–1164. [Google Scholar] [CrossRef]
- Arce, H.; Montero, M.L.; Sáenz, A.; Castaño, V.M. Effect of pH and temperature on the formation of hydroxyapatite at low temperatures by decomposition of a Ca-EDTA complex. Polyhedron 2004, 23, 1897–1901. [Google Scholar] [CrossRef]
- Mao, C.; Li, H.; Cui, F.; Feng, Q. The functionalization of titanium with EDTA to induce biomimetic mineralization of hydroxyapatite. J. Mater. Chem. 1999, 9, 2573–2582. [Google Scholar] [CrossRef]
- Xie, R.; Feng, Z.; Li, S.; Xu, B. EDTA-assisted self-assembly of fluoride-substituted hydroxyapatite coating on enamel substrate. Cryst. Growth Des. 2011, 11, 5206–5214. [Google Scholar] [CrossRef]
- Kalita, S.J.; Verma, S. Nanocrystalline hydroxyapatite bioceramic using microwave radiation: Synthesis and characterization. Mater. Sci. Eng. C 2010, 30, 295–303. [Google Scholar] [CrossRef]
- Hendrickson, H.S. Comparison of the metal-binding properties of nitrilotri(methylenephosphonic) acid and nitrilotriacetic acid: Calcium(II), nickel(II,), iron(III), and thorium(IV) complexes. Anal. Chem. 1967, 39, 998–1000. [Google Scholar] [CrossRef]
- Chau, Y.K.; Shiomi, M.T. Complexing properties of nitrilotriacetic acid in the lake environment. Water Air Soil Pollut. 1972, 1, 149–164. [Google Scholar] [CrossRef]
- Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions, Marvin 17.2.6, 2017, ChemAxon. Available online: http://www.chemaxon.com.
- Ahmed, M.A.; Mansour, S.F.; El-Dek, S.I.; Abd-Elwahab, S.M.; Ahmed, M.K. Characterization and annealing performance of calcium phosphate nanoparticles synthesized by co-precipitation method. Ceram. Int. 2014, 40, 12807–12820. [Google Scholar] [CrossRef]
- Singh, S.; Singh, V.; Aggarwal, S.; Mandal, U.K. Synthesis of brushite nanoparticles at different temperatures. Chem. Pap. 2010, 64, 491–498. [Google Scholar] [CrossRef]
- Maity, J.P.; Lin, T.J.; Cheng, H.P.H.; Chen, C.Y.; Reddy, A.S.; Atla, S.B.; Chang, Y.F.; Chen, H.R.; Chen, C.C. Synthesis of brushite particles in reverse microemulsions of the biosurfactant surfactin. Int. J. Mol. Sci. 2011, 12, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Tas, A.C. Brushite (CaHPO4·2H2O) to octacalcium phosphate (Ca8(HPO4)2(PO4)4·5H2O) transformation in DMEM solutions at 36.5 °C. Mater. Sci. Eng. C 2010, 30, 245–254. [Google Scholar] [CrossRef]
- Sauer, G.; Zunic, W.; Durig, J.; Wuthier, R. Fourier transform Raman spectroscopy of synthetic and biological calcium phosphates. Calcif. Tissue Int. 1994, 54, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Petrov, I.; Šoptrajanov, B.; Fuson, N.; Lawson, J.R. Infra-red investigation of dicalcium phosphates. Spectrochim. Acta Part A Mol. Spectrosc. 1967, 23, 2637–2646. [Google Scholar] [CrossRef]
- Beevers, C.A. The crystal structure of dicalcium phosphate dihydrate, CaHPO4·2H2O. Acta Crystallogr. 1958, 11, 273–277. [Google Scholar] [CrossRef]
- Suguna, K.; Sekar, C. Role of Strontium on the Crystallization of calcium hydrogen phosphate dihydrate (CHPD). J. Miner. Mater. Charact. Eng. 2011, 10, 625–636. [Google Scholar] [CrossRef]
- Zhou, S.; Su, Q.; Li, X.; Weng, J. A novel in situ synthesis of dicalcium phosphate dehydrate nanocrystals in biodegradable polymer matrix. Mater. Sci. Eng. A 2006, 430, 341–345. [Google Scholar] [CrossRef]
- Mondal, S.; Mondal, B.; Dey, A.; Mukhopadhyay, S.S. Studies on processing and characterization of hydroxyapatite biomaterials from different bio wastes. J. Miner. Mater. Charact. Eng. 2012, 11, 55–67. [Google Scholar] [CrossRef]
- Cüneyt Tas, A. Synthesis of biomimetic Ca-hydroxyapatite powders at 37 °C in synthetic body fluids. Biomaterials 2000, 21, 1429–1438. [Google Scholar] [CrossRef]
- Alobeedallah, H.; Ellis, J.L.; Rohanizadeh, R.; Coster, H.; Dehghani, F. Preparation of nanostructured hydroxyapatite in organic solvents for clinical applications. Trends Biomater. Artif. Organs 2011, 25, 12–19. [Google Scholar]
- Sofwan, A.; Alqap, F.; Sopyan, I. Low temperature hydrothermal synthesis of calcium phosphate ceramics: Effect of excess Ca precursor on phase behaviour. Indian J. Chem. 2009, 48A, 1492–1500. [Google Scholar]
- Barbosa, C.C.; Barrias, C.C.; Barbosa, M.A. Preparation and characterisation of calcium-phosphate porous microspheres with a uniform size for biomedical applications. J. Mater. Sci. Mater. Med. 2006, 17, 455–463. [Google Scholar] [CrossRef]
- Sanosh, K.P.; Chu, M.C.; Balakrishnan, A.; Lee, Y.J.; Kim, T.N.; Cho, S.J. Synthesis of nano hydroxyapatite powder that simulate teeth particle morphology and composition. Curr. Appl. Phys. 2009, 9, 1459–1462. [Google Scholar] [CrossRef]
- Choi, D.; Marra, K.G.; Kumta, P.N. Chemical synthesis of hydroxyapatite/poly(ε-caprolactone) composites. Mater. Res. Bull. 2004, 39, 417–432. [Google Scholar] [CrossRef]
- Thomas, S. Online Programme zur Spektroskopie. 2012. Available online: http://www.spec-online.de.
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable prenucleation calcium carbonate clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Kellermeier, M.; Gale, J.D.; Bergström, L.; Cölfen, H. Pre-nucleation clusters as solute precursors in crystallisation. Chem. Soc. Rev. 2014, 43, 2348–2371. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Oliveira, C.; Rocha, F. The different phases in the precipitation of dicalcium phosphate dihydrate. J. Cryst. Growth 2003, 252, 599–611. [Google Scholar] [CrossRef]
- Taubert, A.; Balischewski, C.; Hentrich, D.; Elschner, T.; Eidner, S.; Günter, C.; Behrens, K.; Heinze, T. Water-soluble cellulose derivatives are sustainable additives for biomimetic calcium phosphate mineralization. Inorganics 2016, 4, 33. [Google Scholar] [CrossRef]
- Casse, O.; Colombani, O.; Kita-Tokarczyk, K.; Muller, A.H.E.; Meier, W.; Taubert, A.; Müller, A.H.E. Calcium phosphate mineralization beneath monolayers of poly(n-butylacrylate)-block-poly(acrylic acid) block copolymers. Faraday Discuss. 2008, 139, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chu, B.; Hsiao, B.S. Mineralization of hydroxyapatite in electrospun nanofibrous poly(l-lactic acid) scaffolds. J. Biomed. Mater. Res. Part A 2006, 79A, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Taubert, A.; Glasser, G.; Palms, D. Kinetics and particle formation mechanism of zinc oxide particles in polymer-controlled precipitation from aqueous solution. Langmuir 2002, 18, 4488–4494. [Google Scholar] [CrossRef]
- Taubert, A.; Palms, D.; Weiss, Ö.; Piccini, M.-T.; Batchelder, D.N. Polymer-assisted control of particle morphology and particle size of zinc oxide precipitated from aqueous solution. Chem. Mater. 2002, 14, 2594–2601. [Google Scholar] [CrossRef]
- Taubert, A.; Kübel, C.; Martin, D.C. Polymer-induced microstructure variation in zinc oxide crystals precipitated from aqueous solution. J. Phys. Chem. B 2003, 107, 2660–2666. [Google Scholar] [CrossRef]
- Lee-Thorp, J.P.; Shtukenberg, A.G.; Kohn, R.V. Effect of step anisotropy on crystal growth inhibition by immobile impurity stoppers. Cryst. Growth Des. 2017, 17, 5474–5487. [Google Scholar] [CrossRef]
- Salama, A.; Neumann, M.; Günter, C.; Taubert, A. Ionic liquid-assisted formation of cellulose/calcium phosphate hybrid materials. Beilstein J. Nanotechnol. 2014, 5, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Shkilnyy, A.; Schöne, S.; Rumplasch, C.; Uhlmann, A.; Hedderich, A.; Günter, C.; Taubert, A. Calcium phosphate mineralization with linear poly(ethylene imine): A time-resolved study. Colloid Polym. Sci. 2011, 289, 881–888. [Google Scholar] [CrossRef]
- Hentrich, D.; Junginger, M.; Bruns, M.; Börner, H.G.; Brandt, J.; Brezesinski, G.; Taubert, A. Interface-controlled calcium phosphate mineralization: Effect of oligo(aspartic acid)-rich interfaces. CrystEngComm 2015, 17, 6901–6913. [Google Scholar] [CrossRef]
- Hentrich, D.; Taabache, S.; Brezesinski, G.; Lange, N.; Unger, W.; Kübel, C.; Bertin, A.; Taubert, A. A Dendritic amphiphile for efficient control of biomimetic calcium phosphate mineralization. Macromol. Biosci. 2017, 17, 1600524. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, L. Phosphorylated osteopontin peptides inhibit crystallization by resisting the aggregation of calcium phosphate nanoparticles. CrystEngComm 2012, 14, 8037–8043. [Google Scholar] [CrossRef]
- Jiang, S.; Pan, H.; Chen, Y.; Xu, X.; Tang, R. Amorphous calcium phosphate phase-mediated crystal nucleation kinetics and pathway. Faraday Discuss. 2015, 179, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Montanari, G.; Rodriguez-Blanco, J.D.; Bovet, N.; Stipp, S.L.S.; Tobler, D.J. Impact of citrate ions on the nucleation and growth of anhydrous CaCO3. Cryst. Growth Des. 2017, 17, 5269–5275. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Vessel | Ca(NO3)2 | NTA or EDTA | (NH4)2HPO4 | Solution after Mixing | ||||
|---|---|---|---|---|---|---|---|---|
| c (mM) | V (mL) | c (mM) | m (g) | c (mM) | V (mL) | c(Ca), c(Pi) (mM) | V (mL) | |
| 1 | 40 | 50 | 0 | 0 a,b | - | - | 20 | 100 |
| A | - | - | - | - | 40 | 50 | ||
| 2 | 40 | 50 | 5 | 0.138 a 0.186 b | - | - | 20 | 100 |
| B | - | - | - | - | 40 | 50 | ||
| 3 | 40 | 50 | 10 | 0.275 a 0.372 b | - | - | 20 | 100 |
| C | - | - | - | - | 40 | 50 | ||
| 4 | 40 | 50 | 15 | 0.413 a 0.558 b | - | - | 20 | 100 |
| D | - | - | - | - | 40 | 50 | ||
| Reaction Vessel | Ca(NO3)2 | NTA or EDTA | (NH4)2HPO4 | Solution after Mixing | ||||
|---|---|---|---|---|---|---|---|---|
| c (mM) | V (mL) | c (mM) | M (g) | c (mM) | V (mL) | c(Ca, Pi) (mM) | V (mL) | |
| 1 | 40 | 15 | 0 | 0 a,b | - | - | 20 | 30 |
| A | - | - | - | - | 40 | 15 | ||
| 2 | 40 | 15 | 5 | 0.041 a 0.056 b | - | - | 20 | 30 |
| B | - | - | - | - | 40 | 15 | ||
| 3 | 40 | 15 | 10 | 0.080 a 0.112 b | - | - | 20 | 30 |
| C | - | - | - | - | 40 | 15 | ||
| 4 | 40 | 15 | 15 | 0.124 a 0.168 b | - | - | 20 | 30 |
| D | - | - | - | - | 40 | 15 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hentrich, D.; Tauer, K.; Espanol, M.; Ginebra, M.-P.; Taubert, A. EDTA and NTA Effectively Tune the Mineralization of Calcium Phosphate from Bulk Aqueous Solution. Biomimetics 2017, 2, 24. https://doi.org/10.3390/biomimetics2040024
Hentrich D, Tauer K, Espanol M, Ginebra M-P, Taubert A. EDTA and NTA Effectively Tune the Mineralization of Calcium Phosphate from Bulk Aqueous Solution. Biomimetics. 2017; 2(4):24. https://doi.org/10.3390/biomimetics2040024
Chicago/Turabian StyleHentrich, Doreen, Klaus Tauer, Montserrat Espanol, Maria-Pau Ginebra, and Andreas Taubert. 2017. "EDTA and NTA Effectively Tune the Mineralization of Calcium Phosphate from Bulk Aqueous Solution" Biomimetics 2, no. 4: 24. https://doi.org/10.3390/biomimetics2040024
APA StyleHentrich, D., Tauer, K., Espanol, M., Ginebra, M.-P., & Taubert, A. (2017). EDTA and NTA Effectively Tune the Mineralization of Calcium Phosphate from Bulk Aqueous Solution. Biomimetics, 2(4), 24. https://doi.org/10.3390/biomimetics2040024