1. Introduction
Polymethyl methacrylate (PMMA) has been widely used as a thermoplastic material in oral medicine since the 1940s for applications involving denture bases, temporary crowns, denture re-lining and restorations, cleft palate obturators, and molds for treatment planning [
1]. Although PMMA has many advantages as a biomaterial, such as being inexpensive, light weight and bio-compatible, its low impact resistance and performance defects (e.g., easily fatigues) greatly limit its widespread application [
2]. As a denture base, it can easily be coated by plaque, such as Streptococcus mutans and Candida albicans [
3] in the complex microbial environment of the oral cavity to form a plaque biofilm. Thus can lead to dental caries, denture stomatitis and other oral mucosal diseases [
4]: these performance limitations prevent PMMA from being an ideal denture base.
A variety of nanomaterials, including nanoparticles, nanosheets, and nanotubes, have been incorporated into PMMA to improve its physical and chemical properties [
5]. However, the ability of nanomaterials to improve these properties depends on the material type, size, content, surface charge, and matrix–matrix reaction [
6]. Zbigniew Raszewski et al. [
7] added nano-silica (SiO
2) to thermally cured PMMA and found that the surface hardness and elastic modulus of the PMMA were improved compared to PMMA alone, with this effect strongly dependent on the physical orientation of the filler particles and the capacity to adhere to the resin matrix. However, the flexural strength of PMMA was negatively affected due to the appearance of agglomeration. In addition, Reem Abualsaud [
8] et al. showed that a strong bond was formed between the SiO
2 and PMMA after salinization, which enhanced the interfacial shear strength between the nanomaterial and the matrix and increased the flexural strength of the composite PMMA compared to the untreated PMMA. Mohanmmed M Gad [
9] et al. used zirconia nanoparticles (ZrO
2NPs) treated with a silane coupling agent as the base material and found that the flexural strength of PMMA was enhanced upon the addition of the nanomaterials. However, when the concentration of ZrO
2NPs increased to 5% and 7.5%, agglomeration of the nanoparticles occurred, resulting in a significant decrease in the compressive strength of the PMMA base material.
The poor antibacterial properties of PMMA significantly limit its application as a prosthetic material in the oral cavity. Bacterial biofilm formation in the oral cavity often leads to the failure of the prosthetic over time [
10]. In addition, the growth of bacteria on PMMA can lead to oral infections that can progress into systemic diseases [
11,
12]. Some studies have shown that more than 60% of patients with Candida-associated stomatitis are due to denture restoration [
13]. To overcome these shortcomings, antibacterial drugs and materials have been introduced into denture base materials. Rong Song et al. [
14] reported that the antibacterial activity of PMMA was positively correlated with bacterial growth inhibition. Rinaldi et al. [
15] modified PMMA with citrate-capped silver nanoparticles, and when the concentration of composite nanoparticles was 3.5 wt%, the area of Candida biofilm decreased from 90% to 6%. Silver nanoparticles are known to have broad-spectrum antibacterial activity. Even at low concentrations, silver nanoparticles can inhibit the growth of Streptococcus mutans, Staphylococcus aureus, Candida, and other microorganisms by selectively destroying cell membranes, producing reactive oxygen species that ultimately destroying DNA structure. Although significant progress has been made in mechanical properties and antibacterial properties in the past, improving the mechanical and antibacterial properties of ideal denture base resins has significant implications in oral medicine applications and beyond.
Hexagonal boron nitride (h-BN) is a two-dimensional nanomaterial that has a similar structure to graphene and has excellent mechanical, chemical, and physical properties. Macroscopically, h-BN is a white powder compared with black graphene which is conducive to its application in stomatology [
16,
17,
18]. BNNPs (boron nitride nanoplatelets), a newly developed nanomaterial, was first investigated in some studies for use as a reinforcement material for dental applications, for which its white color and biocompatibility make it suitable [
19]. One study reported on the mechanical properties regarding self-cured acrylic polymethyl methacrylate reinforced with hexagonal boron nitride and stabilized zirconia nanopowders [
20]. At the same time, silver nanoparticles (AgNPs) are widely used in many biomedical fields because of their broad-spectrum antibacterial properties, excellent thermal stability and high biocompatibility [
21,
22,
23].
In this work, AgNPs were self-assembled on h-BN nanosheets (h-BNNs) to prepare a two-dimensional zero-dimensional nanocomposite system comprising h-BN nanosheets and AgNPs (h-BN/AgNPs). Then, a series of h-BN/AgNPs PMMA nanocomposites (hAP) with varying concentrations of h-BNNs/AgNPs were prepared after incorporating them into PMMA. Based on the systematic evaluation of the basic structure of hAP, the mechanical properties and antibacterial properties of hAP were further studied. We expect that the mechanical properties and antibacterial properties of PMMA can be synergistically improved through the addition of the composite. We will further elucidate the interaction mechanism between the composite and the matrix.
2. Materials and Methods
The h-BNNs/AgNPs nanocomposites were prepared by in situ composite methods. 0.2 g of h-BN (Sigma Chemical Co., St. Louis, MO, USA) powder was dispersed in 100 mL of 50% ethanol solution (Damao Chemical Reagent Factory, Tianjin, China) and ultrasonicated for 10 h to obtain a large amount of exfoliated h-BN. The reduction reaction was carried out with AgNO
3 solution under the action of the reducing agent N, N-dimethylformamide (DMF) (Fuyu Fine Chemical Co. Ltd., Tianjin, China), and the ultraviolet light with λ = 185 nm was assisted irradiation for 15 min to accelerate the growth of metal nanoparticles. The grown silver nanoparticles are embedded in the layered h-BN nanosheets (
Figure 1) to form h-BNNs/AgNPs composites, and the color of the solution changes from milky white to light reddish-brown.
The composites were vacuum dried after standing for 2 h and then subjected to characterization testing. The structure and phase composition of the samples were characterized by an X-ray diffractometer (XRD, Smart APEX II, BRUKER, Bremen, German). The Raman Spectra Determination was performed with a Raman microscope (LabRAM HR Evolution, HORIBA Jobin Yvon S.A.S, Paris, France) under an Olympus microscope, with a 532 nm (6 mV) laser, which obtained by scanning 32 times in two seconds. Absorption spectra of h-BNNs dispersions and h-BNNs/AgNPs were recorded at 200–800 nm with a UV spectrophotometer (Specord 50PLUS, Analytik, Jena, Germany). AFM samples were obtained by pipetting an aqueous suspension of sonicated h-BNNs/AgNPs samples onto a smooth mica carrier sheet. The suspension was evenly spin-coated on the mica substrate at 6000 rpm. Atomic force microscopy images (AFM) were acquired using an atomic force microscope (Multimode8, BRUKER, Billerica, MA, USA). For AFM analysis, a silicon tip on a nitride rod was used with ScanAsyst-air contact mode (resonance frequency 50/90 kHz).
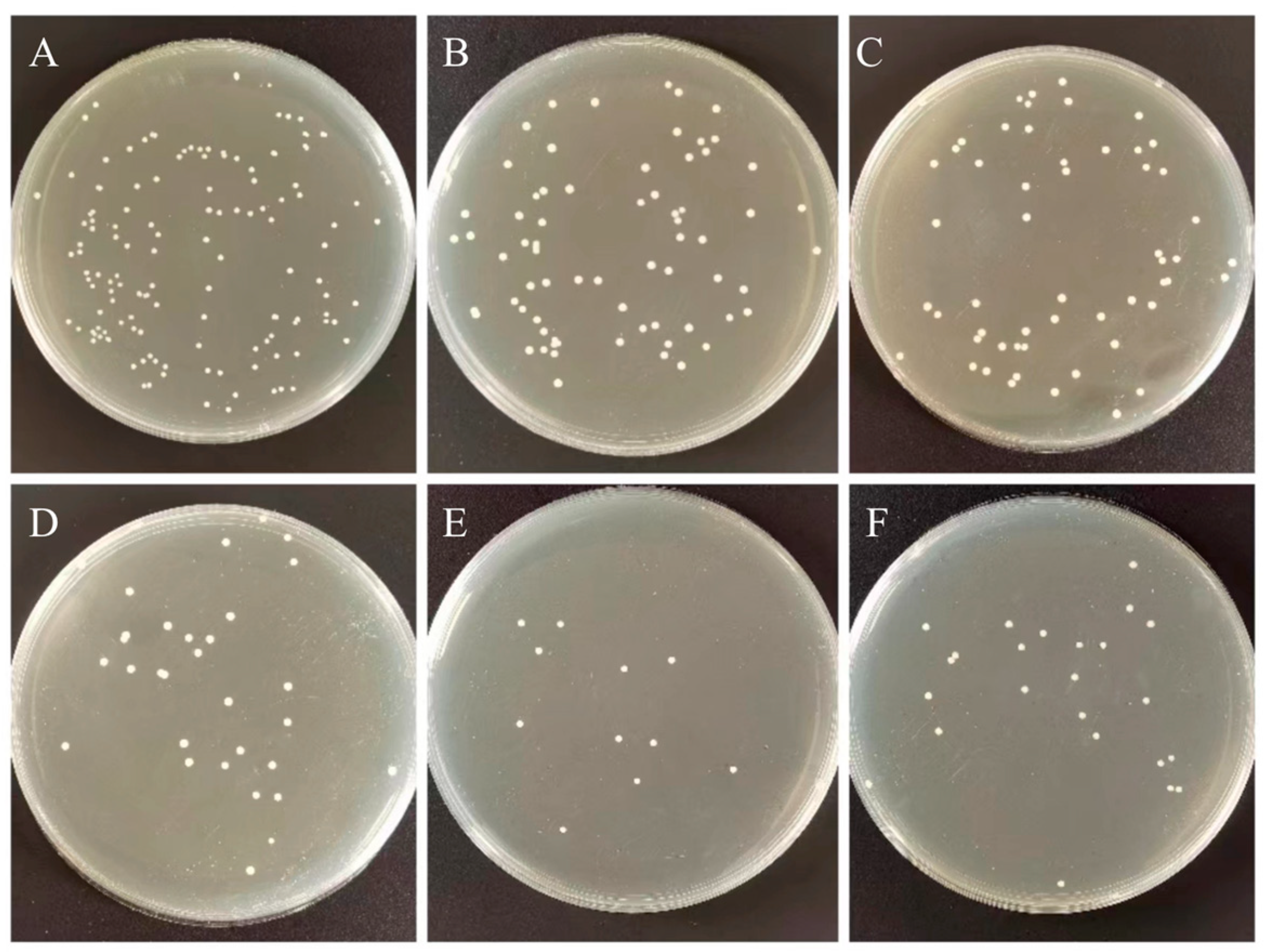
The samples were grouped according to the h-BNNs/AgNPs nanocomposites with different mass fractions, which were hAP containing 0, 0.2, 0.6, 1.0, 1.4, 1.8 wt% h-BNNs/AgNPs in sequence. The compressive strength test (CS) of PMMA is carried out in accordance with the International Organization for Standardization ISO 20795-1 standard. The samples were made in cylindrical metal molds (4 mm diameter × 6 mm height). After preparation, they were placed into distilled water at 37 °C for 24 h for a compression test. The samples for compression testing were loaded on a universal testing machine (R Controller, Testresources, Shakopee, MN, USA) with a crosshead speed of 1 mm/min. The fracture surface of the sample was sprayed with gold, and was observed with a scanning electron microscope (SEM, jsm-5600lv, jeol, Tokyo, Japan).
For flexural strength testing, samples with dimensions (65 mm long × 10 mm wide × 3.3 mm thick) were made according to the international standard ISO 20795-1. Six groups of samples were prepared in total. The samples were stored in deionized water for 24 h, they were subjected to a three-point bending test on a universal testing machine with a transverse head speed of 0.5 mm/min. The samples were placed on rollers at a distance of 15 mm from the center, the entire unit was installed under the universal testing machine, and the stress-applying rods were fixed on the upper part. The center of the sample was made to coincide with the center of the distance between the two rollers. A load was applied with a “T” bar in the center of the specimen until a fracture occurred, at which point the maximum force (F) required to produce a specimen fracture was recorded.
Cylindrical samples with dimensions (4 mm diameter × 6 mm height) were used and, after drying, stored in test tubes at 37 ± 2 °C, with each group having five samples. After 24 h, samples were removed and weighed to an accuracy of 0.0001 g using an analytical balance. The 24 h drying cycle was repeated until a constant mass (m1) was obtained (mass change less than ±0.001 mg). Specimens from each group were immersed in test tubes filled with distilled water, wrapped in aluminum foil to avoid light exposure, and placed in an incubator at 37 ± 1 °C. Sample weights were recorded every 24 h until constant weight (m2) was reached after 7 d. Every 24 h, the specimens were removed from the solution, wiped with a paper towel to remove excess solution, and returned to the solution immediately after weighing. At the end of the soaking period, the specimens were dried as previously described until the specimens reached a constant mass (m3). We then calculated the solubility and water absorption values of the samples.
The sample surface’s static contact angle (CA) was measured using a contact angle meter (SZ-CAMB, Shanghai, China) at room temperature. During the test, the test liquid was placed on the sample surface using a standard microsyringe, and a camera captured the image. In order to obtain an accurate CA value, the contact angle was determined using the ring method, five different positions on the sample surface were measured, and the average value was taken as the test result.
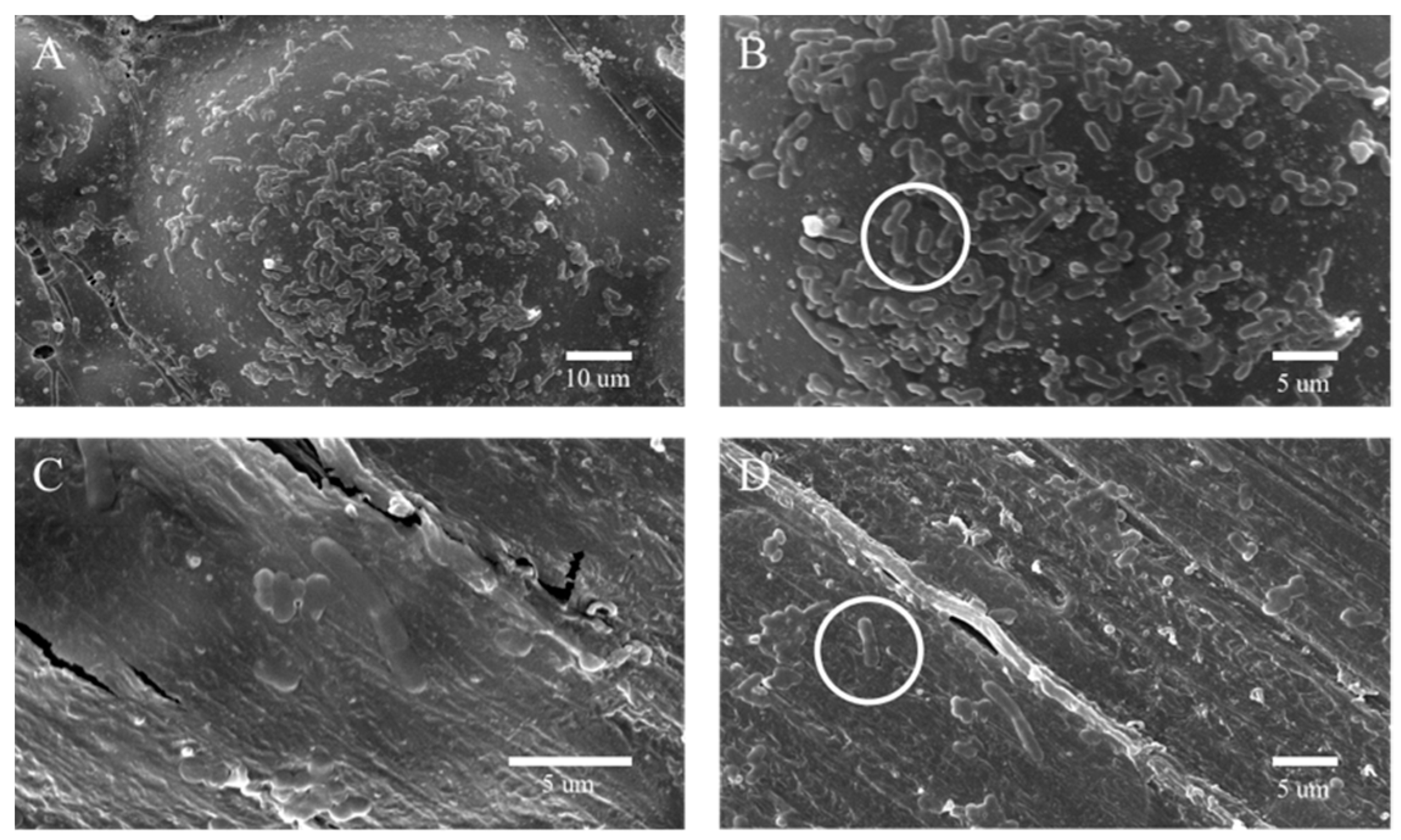
The bacterial plate counting method evaluated the antibacterial ability of the control and experimental groups against Streptococcus mutans (S. mutans, ATCC®700610). Streptococcus mutans were placed in BHI broth and cultured in 5% CO2 incubator at 37 °C for 12 h. The bacterial suspension was diluted to 1:100 with fresh BHI for further use. Each group took one sample (10 mm in diameter × 2 mm in height) for cleaning, drying, and sterilization. We dropped 0.1 mL of Streptococcus mutans suspension at a concentration of 106 CFU/mL on the surface of the plate specimen, and covered with a polyethylene film (30 × 30 mm), so the suspension forms a thin and uniform liquid film. Specimens were incubated at 90% relative humidity at 37 °C for 24 h. The specimen and polyethylene membrane surface was then rinsed with 20 mL PBS. We diluted 0.1 mL of washing solution 100 times with PBS, spread 100 μL of the diluted washing solution evenly on the agar plate, incubated at 37 °C for 24 h, and counted and analyzed the colonies on the plate. The original bacterial solution that did not grow on the surface of PMMA was treated as a blank control group according to the above steps. Measurements were repeated three times for each group. Next, the sample preparation method is the same as the above method: after incubating the bacterial solution and the sample together for 12 h in the incubator, Streptococcus mutans adhered and aggregated on the surface of PMMA. Non-adherent bacteria on the sample surface were washed with PBS.
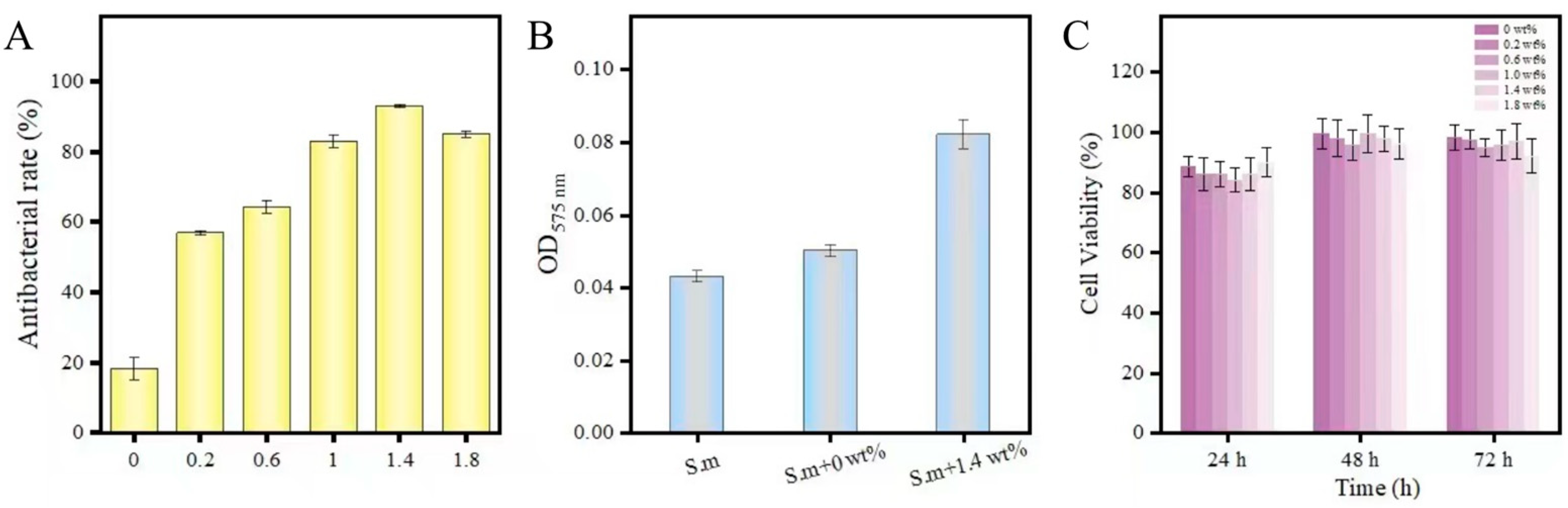
The bacterial sample was fixed with Gluta fixative, and the sample’s surface was sprayed with gold to obtain a scanning electron microscope image of the sample surface. The Nitrotetrazolium Blue chloride (NBT) reduction method measured the intracellular oxidative stress. The bacterial suspension and the samples with the addition of 0 wt% and 1.4 wt% of the complex, respectively, were placed in an incubator at 37 °C for 7 d. Taking 0.1 mL of bacterial solution and add 0.5 mL of NBT, react at 37 °C for 30 min, we added 0.1 mL of hydrochloric acid with a concentration of 1 mol/L to stop the reaction, centrifuged the reacted suspension for 10 min, removed the supernatant, and obtained the precipitate. The cell bodies were treated with dimethyl sulfoxide (DMSO) to extract the reduced NBT from the cells, diluted with PBS. Finally, the absorbance was measured at 575 nm using a UV spectrophotometer.
The in vitro cytotoxicity of 6 groups of PMMA was evaluated by detecting the viability and morphology of fibroblasts (L-929, Shanghai Cell Bank, Chinese Academy of Sciences, Beijing, China). The sample preparation method was the same as before, with 3 replicate groups for each group, a total of 18 samples (10 mm in diameter × 2 mm in height). Samples were stored in deionized water for 24 h and thoroughly sterilized before detection. According to ISO standards, the extraction medium was prepared with DMEM serum-free cell culture medium with a specific surface area/volume ratio of 3 cm2/mL, and the eluate was prepared by incubating at 37 °C and 5% CO2 for 3 d. The cells were seeded in a 96-well cell culture plate with a cell culture medium concentration of 2 × 105 cells/mL well and adhered to the cells after incubation for 24 h. 100 μL of all PMMA extracts and 10 μL of CCK-8; Sigma-Aldrich, Yeasen Biotech Co., Ltd., Shanghai, China) were added to the plate per well. They were incubated for 24, 48, and 72 h in a humidified incubator at 37 °C, 5% CO2, and we measured the absorbance at 490 nm by enzyme labeling. The experiment for each group was independently replicated 3 times.
4. Discussion
In this work, we sought to improve the mechanical properties and antibacterial properties of PMMA by introducing a hybrid system comprising h-BNNs and AgNPs into resin, and the nanocomposite resins were characterized by XRD, Raman and UV-vis spectroscopy, AFM, and other techniques. These methods confirmed the successful synthesis of the h-BNNs/AgNPs system and the addition of the composite to the denture base material [
25].
It is well-known that h-BN and graphite are isostructural [
26], and the exfoliated h-BNNs has a higher specific surface area and bond better with the matrix material. We observed that, as the concentration of the h-BNNs/AgNPs system reached 1 wt% amount, the flexural strength and compressive strength of the PMMA resin was significantly improved over the unmodified PMMA. The compressive strength of the acrylic resin containing 1 wt% h-BNNs/AgNPs was 53.5% higher than that of the unmodified PMMA, while the flexural strength increased by 56.7% over the control. The improved mechanical properties might be related to the good bonding strength between the nanocomposite and matri and uniform distribution of h-BNNs/AgNPs nanocomposite particles throughout the PMMA resin. The nanoparticles were capable of filling in voids surrounding the polymer chains, and when the resins were subjected to external stress, the load could be transferred from the matrix material to the nanofiller. This prevented the propagation of cracks due to the strong interfacial interactions between the nanofillers and the resin matrix. However, when the concentration of the nanocomposite within the polymer resin was high, the mechanical properties decreased due to the agglomeration of the nanomaterials, leading to discontinuities in the resin matrix. In the presence of multiple agglomerates, the stress is concentrated at each agglomeration site, resulting in reduced mechanical properties.
While improving the mechanical properties of the denture base acrylic resin, it is important that the composites used to modify the resin do not deleteriously affect the physical properties of the resin, such as water absorption and solubility. Acrylic resin dentures have a tendency to absorb water, resulting in dimensional denture changes and polymer hydrolysis. The water absorption capacity of a denture base polymer should be less than or equal to 32 μg/mm
3, as recommended by ISO156715. In our studies, the water absorption capacities of the hAP samples were all smaller than the unmodified PMMA, which was attributed to the ability of the nanomaterials to fill in the voids within the polymer resin, endowing the polymer matrix with a more compact and firm structure with lower porosity. The solubility reflects the mass of soluble substances in the polymer. In denture-based acrylic resins, these soluble substances include initiators, activators, plasticizers, and residual monomers [
27]. Previous studies have shown that the water absorption capacity of different types of acrylic resins can reach 10–25 μg/mm
3. In this work, the solubility of the nanocomposite PMMA resins, on average, met the requirements of ISO156715. The results also showed that the water absorption and solubility values were directly related to the concentration of the nanoparticles, as the solubility and water absorption capacity increased as the concentration of the nanoparticles increased. This adverse effect resulted from the aggregation of the nanoparticles at high concentration. Overall, the modified PMMA exhibited a more hydrophobic behavior than the unmodified PMMA.
It has been demonstrated that AgNPs can interact with bacterial cells in different ways [
28,
29]. AgNPs attach to the bacterial cell membrane by bonding to sulfhydryl groups within membrane proteins, causing disruption to the integrity of the membrane. After penetrating the cell membrane, AgNPs damage intracellular DNA and induce the production of ROS, causing oxidative stress responses. In this study, the h-BNNs/AgNPs composites not only significantly improved the mechanical properties of PMMA but also endowed the PMMA with an antibacterial effect. For example, the hAP containing 1.4 wt.% h-BNNs/AgNPs had an AR that was 92.1% higher than pure PMMA in the denture base. The uniformly distribution of the AgNPs throughout the PMMA matrix will allow the PMMA resin to exhibit a sustained antibacterial effect with wear during clinical use. This method of filler addition is ideal for routes over coating modification.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







