1. Introduction
Rolling circle amplification (RCA) occurs naturally to replicate the genomes of certain transposons, bacterial plasmids and viruses having circular genomes [
1]. Rolling circle amplification has also been developed for a number of in vitro applications, such as for isothermal amplification of circularized DNA templates generating amplification products for sequencing, library construction, bioengineering or diagnosis [
2,
3,
4]. Circularized DNA templates are formed upon connecting the ends of linear DNA by ligases such as T4 ligase from Bacteriophage T4 or
Paramecium bursaria chlorella virus (PBCV-1) ligase from
Chlorella virus. For in vitro diagnostics, circular DNA is formed by annealing head-to-tail single-stranded linear 5′-phosphorylated padlock probes of typically 50–200 bp to linear target DNA or RNA and ligase-mediated joining of their ends [
5]. Correct non-mismatched annealing is required for most DNA or RNA ligases what has enabled this method to distinguish single point mutations and polymorphisms at the ligation junction [
6,
7,
8,
9,
10]. In most cases, the circularized padlock probes are extended and amplified by RCA from a start primer or from random hexamer primers (DNA or RNA) that anneal to the circularized padlock probe and initiate the replication reaction by a polymerase with high strand-displacement activity such as Φ29 polymerase or
Bacillus stearothermophilus (Bst) DNA polymerase [
11,
12,
13]. Whereas RCA is a linear amplification technique, a number of modifications in the RCA technique allow also quasi-exponential amplification, such as the ramification or cascade amplification method (RAM), RCA coupled with loop-mediated amplification (RCA-LAMP), or as recently demonstrated by circle-to-circle amplification (C2CA) RCA [
2,
3,
14,
15,
16,
17,
18].
For detection, the linear extended concatemeric single-stranded DNA (ssDNA) amplification products are usually separated as high molecular weight DNA with low migration in matrices such as agarose, polyacrylamide gels or paper, and visualized using fluorescent intercalating dyes such as ethidium bromide or Gel Red [
3]. Alternatively, the ssDNA amplification products are detected using a labeled start primer and/or hybridization of labeled oligonucleotide detection probes and molecular beacons (e.g., labeled with dye/gold, fluorescent, biotinylated, digoxigeninated, or radioactive markers). These labels are then detected by optic, colorimetric, enzymatic, or electrochemical signal amplification reactions in vitro as well as in situ, e.g., in paraffin-embedded tissue slides or in fixed cells for localization and diagnosis [
19,
20,
21,
22,
23,
24]. Fluorescent detection has the advantage that multiple RNA/DNA species can be detected simultaneously using spectrally separable dyes [
8]. A number of fluorescent-labeled nucleotides have also been tested for direct incorporation during RCA and direct or indirect detection by fluorescence resonance energy transfer (FRET) [
25,
26]. However, most of these detection methods for RCA products have relatively low sensitivity (e.g., only one or a few labels are usually present in the detection probes), high background due to non-specific random hybridization and to the presence of endogenous high molecular weight DNA, and are time-consuming due to the long time to hybridize and wash the non-specifically bound probes from the sample.
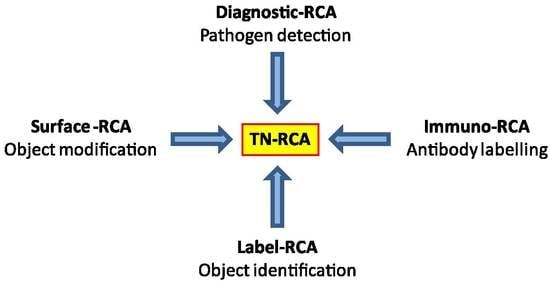
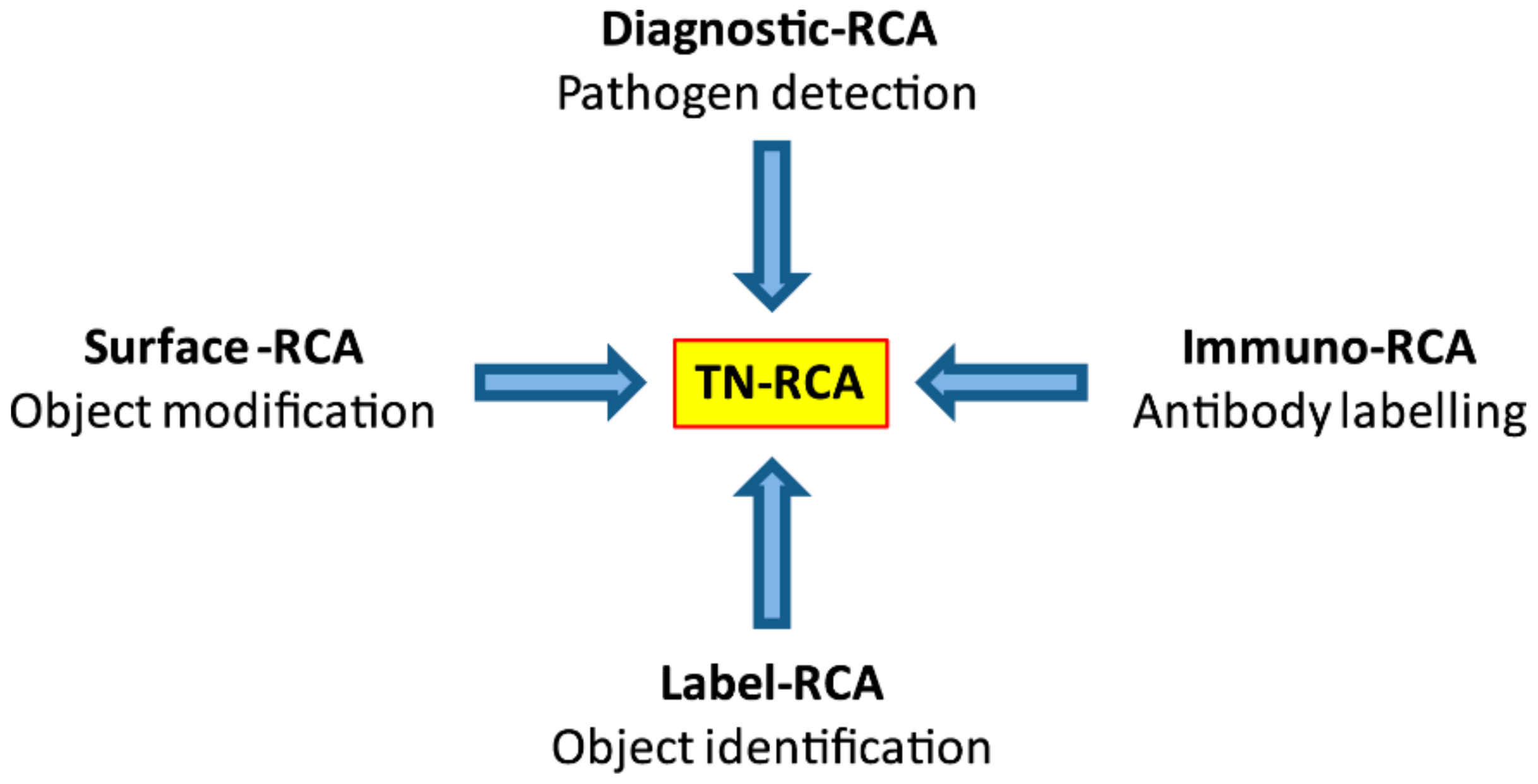
As a diagnostic technique, RCA has been used for detection of DNA or RNA either occurring naturally (genomic or pathogen DNA or RNA) or of synthetic oligonucleotides that have been linked to antibodies (Immuno-RCA) or to microarrays (Surface-RCA) [
2]. For Crimean Congo hemorrhagic fever virus, a negative strand RNA virus, complementary DNA (cDNA) was detected by an in situ RCA technique [
27], and for human immunodeficiency virus (HIV) cDNA, point mutations were detected using RCA [
28]. Recently, a netlike RCA-based point of care test (POCT) was developed for the pH-responsive detection of microRNA [
24]. Using the circle-to-circle amplification (C2CA) method [
17,
18], gold-labeled detection probes and either magnetic beads-based read out or lateral flow on paper strips, drug resistant
Mycobacterium tuberculosis was detected within 75 min [
15,
16]. Although a number of studies have shown the ability of RCA to isothermally detect and diagnose pathogens, the lower rate of linear amplification when compared to polymerase chain reaction (PCR) or other exponential amplification techniques (e.g., helicase-dependent amplification (HDA), recombinase polymerase amplification (RPA), loop-mediated isothermal amplification (LAMP)), and the interference of sample-derived background has so far limited the use of RCA-based techniques in any approved diagnostic clinical or laboratory tests.
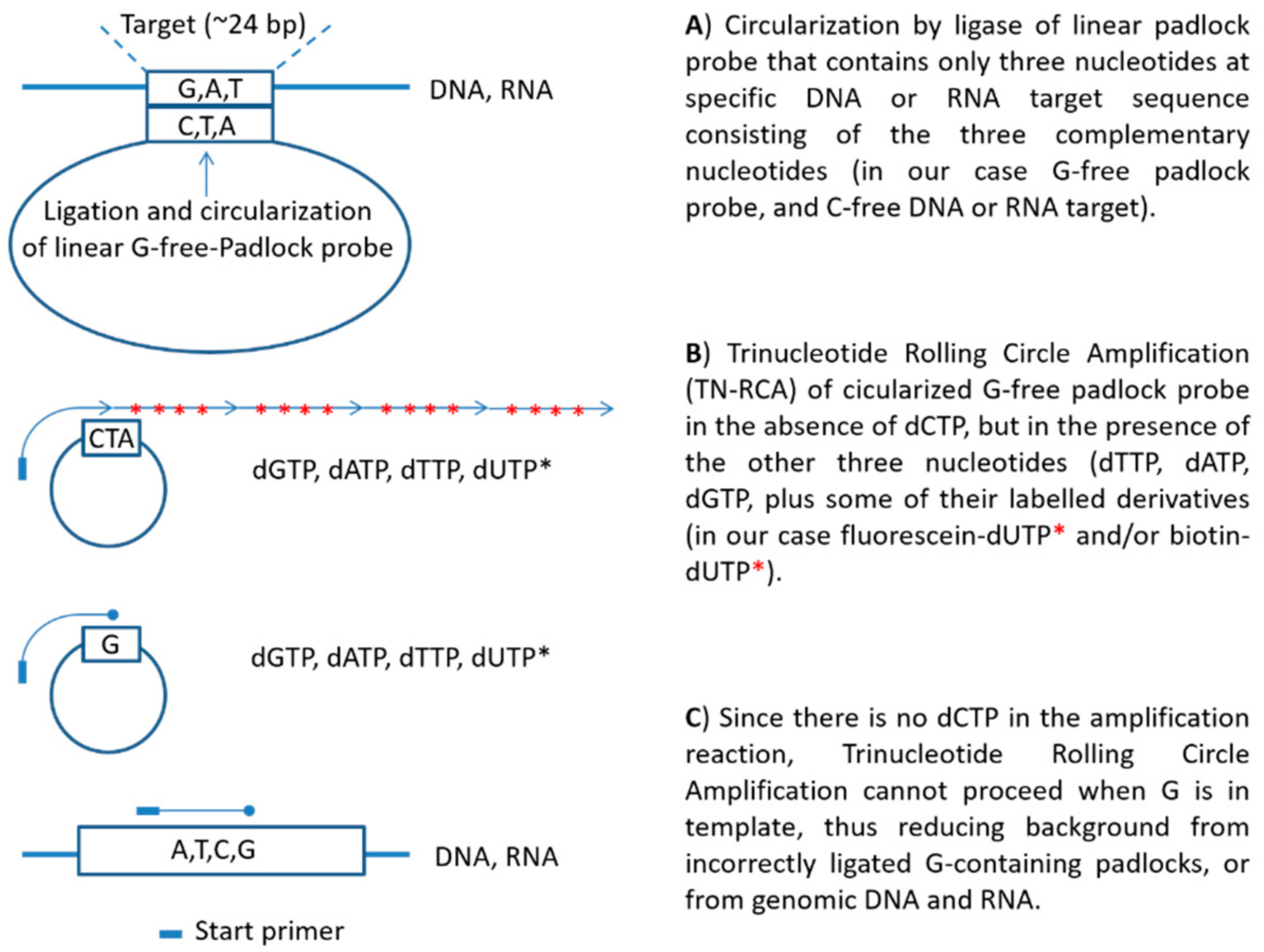
In this study, we developed a novel RCA technique, trinucleotide RCA (TN-RCA), which is based on 5′-phosphorylated padlock probes (~50–200 bp long) that anneal at low temperature sequence-specifically head-to-tail with their ends to specific target sequences lacking one of the four nucleotides (missing nucleotide (MN)) in DNA or RNA (~20–40 bp TN-stretches). The sequences of such padlock probes completely lack the nucleotide complementary to the MN. Since during TN-RCA the MN (as dNTP) is not added to the reaction mixture during polymerization, only the correctly ligated circular padlock probe will amplify minimizing background amplification that may occur at low temperature in the presence of endogenous RNA or DNA (which mostly would require the presence of all four dNTP). Therefore, in the absence of non-specific amplification, various labeled dNTP can be added to the TN-RCA reaction that enables the separation, isolation and detection of the amplified concatemeric linear ssDNA. In the following, TN-RCA is exemplified with a guanosine-free or G-free padlock probe, and for a specific cytidine-free or C-free target sequence in Zika virus DNA and RNA and for human papilloma virus (HPV) DNA.
2. Materials and Methods
2.1. Materials
Zika viral particles from Brazilian Fortaleza and Rio strains (kindly provided by M. Ricciardi and Dr. D. Watkins (Department of Pathology, University of Miami, Miami, FL, USA)) were harvested from Vero cells supernatants, filtered using a 0.4 μm filter, and the viral RNA was isolated using an a QIAamp viral RNA minikit (Qiagen, Germantown, MD, USA). For detection of the presence of Zika virus DNA and for generation of Zika DNA target sequences, reverse transcription polymerase chain reaction (RT-PCR) was performed as described below using these samples. All oligonucleotides were purchased from Sigma (Burlington, MA, USA). HeLa cells (ATCC) were grown in DMEM/10% FCS, 2 mmol/L L-glutamine, 100 μg/mL streptomycin and 100 U penicillin and genomic RNA and DNA was isolated with the Trizol method according to the manufacturers’ protocol (Invitrogen, Waltham, MA, USA).
2.2. Reverse Transcription Polymerase Chain Reaction
Primer pairs for RT-PCR to detect Zika virus were searched using Primer3 software [
29,
30]. Alignments of the sequences from Zika virus strains from Uganda (Genbank NC_012532.1), French Polynesia (Genbank KJ776791.1), and Brazil (Genbank KU497555.1) were further used to select primers that would amplify these three viral strains. A suitable primer pair was identified in a sequence coding for the non-structural protein 1 (NS1), with one mismatch towards the Uganda virus strain. No identical sequences were found in human DNA using BlastN searches [
31]. Alignments with other Flavivirus sequences (Dengue virus 1 to 4, Genbank KT187564.1, KT187558.1, KR296744.1, KP406806.1), Japanese encephalitis virus (Genbank NC_001437.1), yellow fever virus (Genbank NC_002031.1), and the alpha virus Chikungunya virus (Genbank KJ451624.1 and KJ451623.1) showed that these viruses will not be amplified. RT-PCR was performed in a single tube using the Tth DNA polymerase according to the one-step RT-PCR protocol given by the manufacturer (Roche, Basel, Switzerland). Briefly, 5 μL of viral RNA or particle were assembled in reaction buffer with 1 μL of 50 μM of each primer Zikafw and Zikarv: Zikafw: 5’-GCTTGAAATTCGGTTTGAGG-3’; Zikarv: 5’-CTTTCCTGGGCCTTATCTCC-3’.
The reverse transcription (RT) reaction was at 60 °C for 30 min. Then, the samples were heated to 94 °C for 1 min, followed by 40 cycles at 94 °C, 30 s, at 38 °C, 30 s and at 72 °C, 45 s, with a final elongation step at 72 °C for 7 min. The PCR products were separated by a 2% agarose gel, extracted with a gel extraction kit (Qiagen) and sequenced (Genewiz, South Plainfield, NJ, USA) or used for TN-RCA reactions.
2.3. Trinucleotide Rolling Circle Amplification Design
Stretches of 20–30 bp DNA sequences in which cytidine (C) was absent were searched in aligned genomic sequences for Zika virus strains from Uganda (Genbank NC_012532.1), French Polynesia (Genbank KJ776791.1), and Brazil (Genbank KU497555.1). A stretch of 24 bp was identified in a sequence of the nonstructural protein 1 (NS1) partially overlapping with the Zikarv PCR primer, that was C-free and identical in the three viral strains with the exception of one C in the Uganda strain. Interestingly, when the selected target sequence (5’-TGTTGGTATGGAATGGAGATAAGG-3’) was searched using BlastN, 100% homology was found not only with Zika virus, but 95% homology was also found with the Usutu virus, an emerging Flavivirus in Europe [
32]. The 5′-phosphorylated G-free padlock probe was so designed that the 5′-end anneals to the first 12 bp of the positive strand of the target, and the 3′-end to the second 12 bp, so that ligation will occur between the two adenosines (A) in the middle. Two A at the target sequence have been reported to be efficiently ligated both by T4 ligase in DNA/DNA [
33] as well as by SplintR Ligase in DNA/RNA hybrids [
34]. The following oligonucleotides were used as the short synthetic Zika virus target DNA and RNA (bold), and as 5′-phosphorylated Zika virus G-free padlock probes. Zika virus RNA and DNA targets (bold the target sequence): ZTargetupper: 5-TAAAGATGGC
TGTTGGTATGGAATGGAGATAAGGCCCAGGAAAG-3; ZTargetlower: 5-CTTTCCTGGG
CCTTATCTCCATTCCATACCAACAGCCATCTTTA-3; ZRNA: 5-UAAAGAUGGC
UGUUGGUAUGGAAUGGAGAUAAGGCCCAGGAAAG-3.
Padlock probes (bold: ends that anneal to the target sequence; underlined: sequence recognized by the start primer).
Padlock probe a: G-free padlock probe for Zika, GfreeZ (84 bp):
5’-p-TCCATACCAACATTTTTATCTTAACTCACCAACACCATTTTTTCTAATCTCAACCTTACTACACTCTTTTTTCCTTATCTCCAT-3’
Padlock probe b: G-free padlock 74, GfreeZ74 (74 bp):
5’-p-TCCATACCAACATTTTTATCTTAACTCACCAACACCATCTCAACCTTACTACACTCTTTTTTCCTTATCTCCAT-3’
Padlock probe c: G-free padlock 74 one mismatch, GfreeZ1mis (74 bp, mismatched bases is lower case):
5’-p-TCCATACCAACATTTTTATCTTAACTCACCAACACCATCTCAACCTTACTACACTCTTTTTTCCcTATCTCCAT-3
Padlock probe d: G-free padlock 74 two mismatches, GfreeZ2mis (74 bp, mismatched bases are lower case):
5’-p-TCCATACCAtCATTTTTATCTTAACTCACCAACACCATCTCAACCTTACTACACTCTTTTTTCCcTATCTCCAT-3’
Start primer A:
5’-TGGTGTTGGTGAGTTAAG-3’
The calculated melting temperature (Tm) of the non-mismatched 5′-end and 3′-ends that anneal to the target sequence were both 34 °C, which is relatively low due to the absence of G in the sequence. The calculated Tm of the start primer was 49 °C. The start primer did not reveal high homology to human DNA in BlastN searches. A search for self-dimers or hairpins in the G-free padlock probe and the start primer using OligoAnalyzer 3.1 (Integrated DNA Technologies, Inc, Coralville, IA, USA) did not reveal any secondary structure that would be disadvantageous.
2.4. Considerations for Developing Assay Conditions of TN-RCA for Specific Target Sequences
As described in the following, assay design for TN-RCA may need to take into consideration several specific features that depend on the target type (single/double stranded DNA/RNA, length, target sequence). For efficient TN-RCA with genomic DNA targets, the DNA may be cleaved with restriction endonucleases close to the 3′-side of the padlock probe and then denatured and annealed to the padlock as outlined below. Annealing of DNA/DNA will generate B-DNA type helix of 10 bp per helical turn, which can be ligated efficiently by T4 ligase from Bacteriophage T4 and 10 mM ATP in the presence of fresh dithiothreitol (DTT). The efficiency of ligation has been shown to be dependent on certain nucleotides at the ligation junction that may need to be considered [
33]. During amplification by the polymerase cutting the target DNA close to the 3′-side of the padlock probe will facilitate the release of the circular padlock probe from the intertwined genomic DNA and generate a 3′-end for self-priming. Cutting of the target DNA can also be achieved by annealing padlock probes with a T/A mismatch and subsequent cleavage of the adenine in the target sequence by MutY adenine DNA glycosylase. Whole genome amplification and protein nucleic acid (PNA) probes can be used to make the target more accessible for the padlock probes [
35].
For efficient TN-RCA with RNA, SplintR Ligase (NEB, Ipswich, MA, USA), PBCV-1 Ligase from
Chlorella virus, or T4 RNA ligase 2 (Rnl2) is preferentially used [
34,
36]. Annealing of DNA/RNA will generate A-DNA-like structure having intermediate characteristics between the A- and B-DNA-type structure and between 10 and 11.6 bp per helical turn. During amplification by the polymerase, RNases (e.g., RNase H, RNase A, RNase III) may be added what will hydrolyze specifically the RNA in DNA/RNA hybrids thus releasing the circular padlock probe form the intertwined RNA. When T4 ligase needs to be used with an RNA target, the ATP concentration should be reduced to 10 μM, since otherwise inhibitory AppLigase complexes are formed [
33,
37].
Most natural DNA and RNA are devoid of long trinucleotide (TN) stretches (20–200 bp) of sequences that lack one specific nucleotide (missing nucleotide (MN)). However, there are exceptions, such as disease-associated trinucleotide expansions, polyAAA tails of mRNA or as present in some pseudogenes. Trinucleotide rolling circle amplification is also expected to work with these special cases, although specific adaptations may be required. For specific applications, dinucleotide RCA (DN-RCA) or even mononucleotide RCA (MN-RCA) can be envisioned, e.g., for the detection in genomic DNA or RNA of dinucleotide repeats or polyAAA tails, respectively, but the suitability of padlock probes containing only two or even one nucleotide for amplification remains to be determined. In these cases, dinucleotide targets can be detected using padlock probes with complementary dinucleotide sequences at the end for annealing, but trinucleotides sequences in the remaining sequence, thus allowing again TN-RCA. In particular, TN-, DN-, or MN-RCA can be used to detect synthetic oligonucleotides that have been linked to antibodies (immuno-RCA) or to microarrays (surface-RCA) [
2].
In an optional denaturation/renaturation step prior to the circularization step, the target polynucleotide sequence is denatured (e.g., by heat or alkali for about 5 to 10 min) and then renatured by cooling or neutralization buffer, respectively. Alternatively, the single-stranded target polynucleotide sequence is generated by digesting the strand complementary to the target sequence by using nicking enzymes and exonuclease III [
9], or the target DNA is opened by a peptide nucleic acid probe (PNA) [
35]. The requirements of this step depend on whether the target polynucleotide sequence is single-stranded or double-stranded DNA or RNA or has secondary structure. High molecular weight polynucleotides (e.g., genomic DNA) can also be physically (e.g., acoustic shearing, ultrasonication, hydrodynamic shear) or enzymatically (e.g., by non-specific nucleases and T7 endonuclease) fragmented, and/or cut by specific restriction endonucleases or nicking endonucleases preferentially at sites close to the target sequence, and/or made single-stranded by whole genome amplification, before denaturation. RNA can also be fragmented chemically by heat and divalent metal cations (magnesium or zinc), or enzymatically by RNase III digestion [
38], or RNA is converted to cDNA by reverse transcriptase before denaturation and RNase digestion.
2.5. Basic Protocol of TN-RCA Reaction with DNA and G-Free Padlock Probe
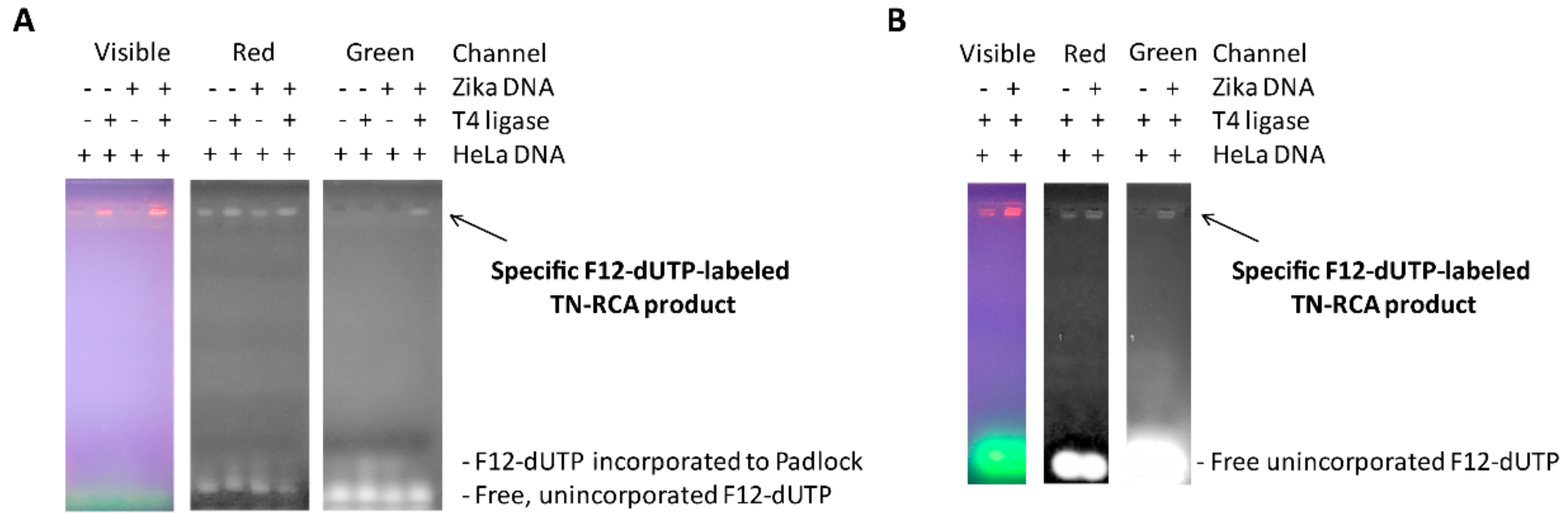
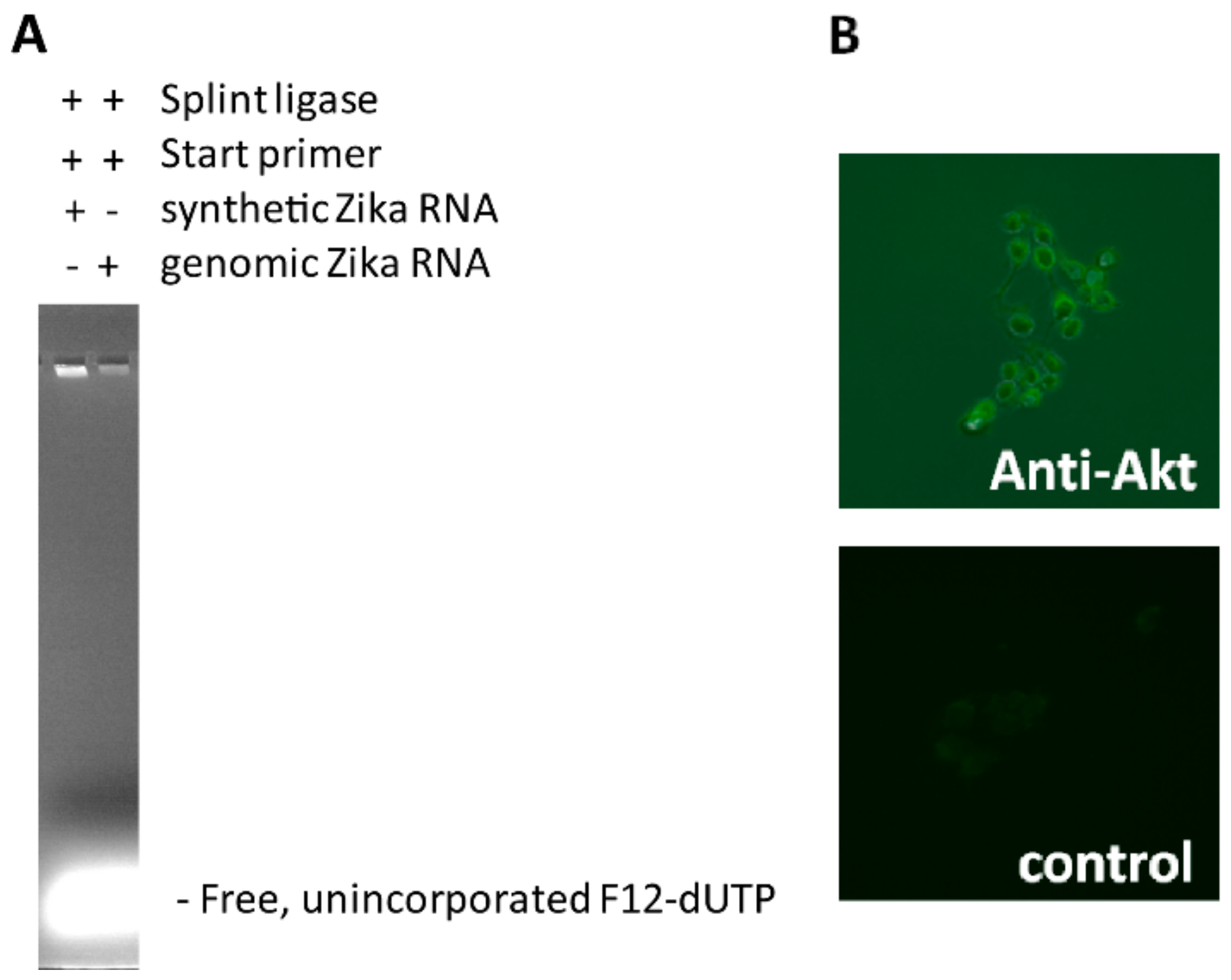
5 μL of Zika virus DNA (18 ng/reaction) and 5 μL of HeLa total genomic DNA (120 ng/reaction) as non-specific human background is denatured for 5–10 min with 10 μL lysis/denaturation solution (400 mM KOH, 5 mM EDTA), and then re-natured with 20 μL Neutralization buffer (300 mM Tris-HCl, 200 mM HCl, prepared by mixing 3 mL of 1M Tris-HCl (pH 7.5) and 2 mL of 1 M HCl with 5 mL of water). The pH in the sample after neutralization was between 7.5 and 7.7 as measured using an Amana-1000-L pH Sensor (Innovative Instrument Inc., Tampa, FL, USA). Then, 10 μL of neutralized DNA is immediately added to the ligation reaction mixture containing 3 μL 10× T4 ligase buffer (NEB), 6 μL 1 μM 5′-phosphorylated G-free padlock probe (Sigma), 2 μL T4 DNA Ligase (NEB) (400,000 U/mL), and to 30 μL ultrapure distilled water (Ambion, Austin, TX, USA), and then incubated at 30 °C for 30 min. To this solution a TN-RCA polymerization mixture is added consisting of 6 μL Φ29 10× reaction buffer (NEB), 1.2 μL of 50 μM start primer (Sigma), 0.6 μL of 10 μg/μL BSA (NEB), 1.2 μL dNTP mix (dATP, dGTP, dTTP, 25 mM each or diluted amounts of this stock), labeled Fluorescein-12-dUTP (various amounts of 1 mM stock, ThermoFisher, Waltham, MA, USA), and/or Biotin-11-dUTP (various amounts of 1 mM stock, ThermoFisher), and 1 μL Φ29 Polymerase (NEB) (2000 U/mL), various amounts of ultrapure distilled water (Ambion) to 60 μL, and then incubated at 30 °C for 0.5–6 h. At the end, 10 μL of 6× gel loading dye is added, the sample mixed and separated by 1.5% agarose gel electrophoresis. The gel is photographed under UV light, and the three channels (visible, red, green) visualized using Adobe Photoshop CS 5.1 software (Adobe, San Jose, CA, USA).
2.6. Basic Protocol for TN-RCA Reaction with RNA and G-Free Padlock Probe
5 μL of Zika virus RNA synthetic template ZRNA (18 ng/reaction) or Zika virus genomic RNA (40–80 ng/reaction) and 5 μL of HeLa total genomic DNA (120 ng/reaction) is optionally denatured for 5–10 min with 10 μL lysis/denaturation solution (400 mM KOH, 5 mM EDTA), and then re-natured with 20 μL Neutralization buffer (300 mM Tris-HCl, 200 mM HCl, prepared by mixing 3 mL of 1M Tris-HCl (pH 7.5) and 2 mL of 1 M HCl with 5 mL of water) (for ssRNA/ssDNA denaturation/renaturation is optional). Then, 10 μL of neutralized RNA is immediately added to the ligation reaction mixture containing 3 μL 10× Splint Ligase buffer (NEB), 6 μL 1 μM 5′-phosphorylated G-free padlock probe (Sigma), 2 μL SplintR Ligase (NEB) (25,000 U/mL), and to 30 μL ultrapure distilled water (Ambion), and then incubated at 30 °C for 30 min. To this solution a TN-RCA polymerization mixture is added consisting of 6 μL Φ29 10× reaction buffer (NEB), 1.2 μL of 50 μM start primer (Sigma), 0.6 μL of 10 μg/μL bovine serum albumin (BSA; NEB), 1.2 μL dNTP mix (dATP, dGTP, dTTP, 25 mM each or diluted amounts of this stock), labeled Fluorescein-12-dUTP (various amounts of 1 mM stock; ThermoFisher), and/or Biotin-11-dUTP (various amounts of 1 mM stock; ThermoFisher), and 1 μL Φ29 Polymerase (NEB; 2000 U/mL), various amounts of ultrapure distilled water (Ambion) to 60 μL, and then incubated at 30 °C for 0.5–6 h. At the end, 10 μL of 6× gel loading dye is added, the sample mixed and separated by 1.5% agarose gel electrophoresis. The gel is photographed under UV light, and the three channels (visible, red, green) visualized using Adobe Photoshop CS 5.1 software.
2.7. Self-Priming TN-RCA in the Absence of Start Primers
For short RNA/DNA targets, the 3′-end of the target oligonucleotide can serve as efficient starting point for self-priming so that no start primer needs to be added. Moreover, since Φ29 polymerase has 3′ to 5′ RNase activity which digests ssRNA [
39], the non-digested RNA remaining annealed to the ligated padlock probe can serve as start site of the TN-RCA reaction, so that even for longer target RNA the start primer is not necessary and can be left away in the reaction mixture. For longer RNA targets digestion can be facilitated by adding specific RNases (e.g., RNase III, RNase H) [
38,
39], whereas longer DNA targets may need to be generated by restriction endonucleases, nickases, whole genome amplification, or dsDNA Fragmentase (NEB). For DNA targets, a self-priming 3′-end can also be generated by annealing padlock probes with a T/A mismatch and subsequent cleavage of the adenine in the target sequence by MutY adenine DNA glycosylase. Accordingly, with DNA, self-priming can be initiated after digesting the non-annealed ssDNA by the 3′ to 5′ exonuclease activity of Φ29 [
40].
2.8. Generation of Circularized Padlock Probes
Circularized padlock probes were generated using the CircLigase ssDNA Ligase kit according to the manufacturers’ protocol (Epicentre, Madison, WI, USA). Briefly, Padlock probes were incubated for 1 h at 60 °C and CircLigase then inactivated for 10 min at 80 °C.
2.9. Digestions with MseI, Exonucleases and RNases
The TN-RCA reaction product was digested with MseI (NEB) (10 U) for 20 min at 37 °C in the presence or absence of cutting primer, Msecutprimer: 5’-TTTATCTTAACTCACCAACT-3’ (underlined: MseI recognition site), and the enzyme subsequently inactivated at 65 °C for 20 min. Lambda exonuclease (NEB) (0.5 U), exonuclease III (NEB) (20 U), exonuclease VIII (NEB) (2 U), T7 exonuclease (2 U), RNase H (NEB) (1 U), RNase A (Thermo Scientific, Waltham, MA, USA) (10 U), RNase A/T1 Mix (Thermo Scientific) (1 U), RNase T1 (Invitrogen/Ambion) (1 U), ShortCut RNase III (NEB) (0.4 U) were added to the TN-RCA reaction mixture after the ligation step was completed, or at various times during the TN-RCA reaction. In some experiments, ShortCut RNase III (NEB) (0.4 U) was also added before the ligation step to fragment the target genomic Zika RNA. Random hexamers (Exo-Resistant Random Primer; 0.2 μL of 500 μM stock; Thermo Scientific) also was added after the ligation step.
2.10. Incorporation of dUTP and Digestion with Uracil DNA Glycosylase
1 μL of Deoxy-UTP (dUTP) from various working stock dilutions (1, 0.1, 0.01 mM) (Roche) was added to the TN-RCA reaction mixture, and Uracil-DNA glycosylase (Roche) (1 U) was added at various time points with/without endonuclease IV (NEB) (10 U) as outlined in the Figure legends.
2.11. Detection Methods
2.11.1. Agarose Gel
As described above high molecular weight TN-RCA reaction products are efficiently separated from background signals by 1.5–1.7% agarose gels, and when labeled fluorescein-12-dUTP is added to the reaction, no ethidium bromide or Gel Red is necessary to visualize the products in the gel. Images of agarose gels without ethidium bromide or Gel Red are acquired using the Epi Blue setting by an Azure Biosystems C200 Imaging system (Dublin, CA, USA).
2.11.2. Microtiter Plate
60 μL of the biotinylated TN-RCA reaction is added to the well of a Neutravidin microtiter plate (Pierce Neutravidin Coated High Binding Capacity (HBC) White 96-well Plates with SuperBlock blocking buffer) and incubated for 20 min. The well is washed 3 times with 200 μL PBST (1 tablet PBS (Invitrogen) in 100 mL H
2O and 100 μL (0.1%) of Tween-20, filtered with a 0.2-µm filter) and blocked with 200 μL PBST/1% BSA for 10 min. Detection is done enzymatically with the Opti-4CN substrate kit (Bio-Rad, Hercules, CA, USA) by adding 150 μL of 1/1000 dilution of Blotting grade avidin-horseradish peroxidase (HRP) in antibody dilution buffer (PBST/1% BSA) for 20 min, washed with 200 μL PBST for 3× 5 min, and detected by adding 0.2 mL of Opti-4CN substrate per 10 mL diluents (mixed one part of Opti-4CN diluent with 9 parts H
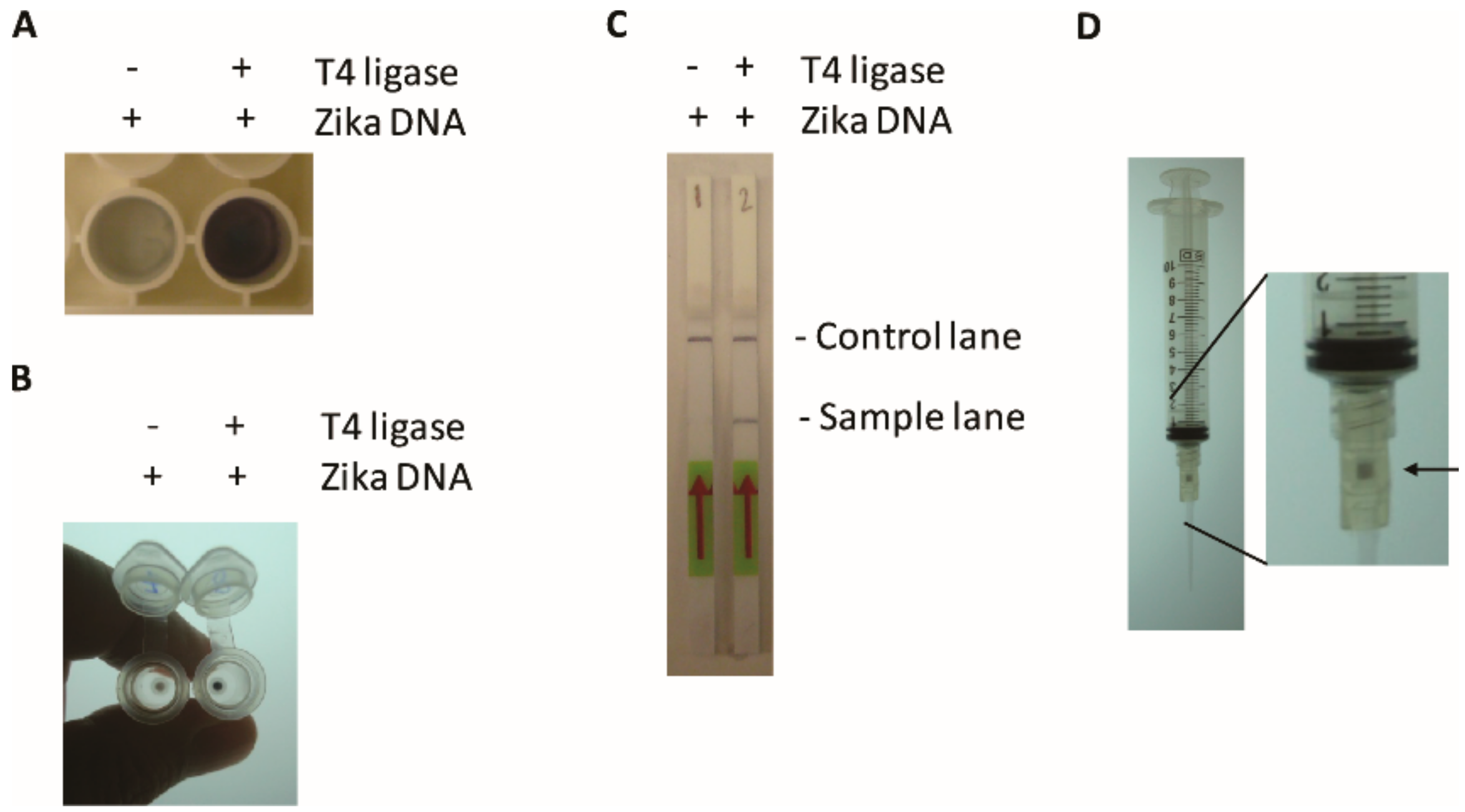
2O). The plate is incubated for up to 30 min with shaking and documented. Alternatively, instead of the microtiter plate, the sample can be dot blotted onto nitrocellulose membranes and air dried and detected as above using the Opti-4CN kit according to the manufacturers’ protocol (Bio-Rad). This method gives no background when the ligase is omitted in the TN-RCA reaction, but it gives a very low background without target DNA/RNA and TN-RCA when the basic protocol is used. This background is most likely due to the extension of the start primer along the linear padlock probe, which leads to incorporation of some biotin-11-dUTP up to the end of the unligated padlock probe (
Figure S1). In addition, with synthetic short target RNA/DNA, signals may be generated by annealing the target to padlock probe and extension to the end of the linear padlock probe. As discussed below, background can be avoided by leaving away the start primer and use instead self-priming, e.g., after digestion of the target with various RNases, that digest the non-annealed target generating a free 3′-end for priming.
2.11.3. DNA Affinity Column
Separation using a DNA affinity column is essentially based on the Monarch DNA PCR and DNA cleanup kit (Monarch, NEB) whereas detection is based on the Opti-4CN detection kit (Bio-Rad). 60 μL of the 11-biotin-dUTP labeled TN-RCA reaction is added to 420 μL of binding buffer, then loaded on spin column and centrifuged for 1 min. The column is washed 2× with 200 μL wash buffer and spun for 1 min in between. Then, 200 μL of 1/1000 dilution of blotting grade Avidin-HPR in antibody dilution buffer (PBST/1% BSA) from the Opti-4CN Substrate Kit (Bio-Rad) is added, centrifuged for one minute, washed 2× with 200 μL binding buffer (Monarch, NEB) (spin 1 min in between). Then, one part of Opti-4CN diluent with 9 parts H2O is prepared and 0.2 mL of Opti-4CN substrate per 10 mL of diluents is mixed, 200 μL added to the spin column and incubated for 5–30 min. As alternative to the centrifugation step that requires a centrifuge and electricity, the filter is attached to a 10 mL syringe, and the same procedure is performed by manually generating a vacuum and sucking all the solutions into the syringe.
2.11.4. Lateral Flow Assay
12-Fluorescein-dUTP- and 11-Biotin-dUTP- labeled samples (1–10 μL of TN-RCA reaction) were spotted on the Hybridetect Dipstick, processed as described in the manufacturers’ protocol (Milenia Biotec, Giessen, Germany), and photographed. Alternatively, 12-Fluorescein-dUTP labeled samples (1–10 μL of TN-RCA reaction) were spotted on the Hybridetect Dipstick, processed as described in the manufacturers protocol (Milenia Biotec) but with the addition of 1 μL of 50 μM 5′-biotin-labeled detection probe able to hybridize to the TN-RCA product (Biotin-5’-CTCAACCTTACTACACTC-3’) to the running buffer, and photographed. The band intensity was assessed using the AlphaEaseFC Software (AlphaInotech, San Leandro, CA, USA).
2.12. In Situ TN-RCA
The oligonucleotide containing the Zika virus target sequence (bold) (ZTargetamine: 5’-amine-C6-MMT-TAAAGATGGCTGTTGGTATGGAATGGAGATAAGGCCCAGGAAAG-3’) was covalently attached to an IgG secondary antibody (goat anti-rabbit IgG, whole molecule; Sigma), using an oligonucleotide-conjugation kit according to the manufacturers’ procedure (Abcam, Cambridge, MA, USA). HeLa cells were grown in Falcon 8-well cell culture slides, fixed with 10% formalin/PBS, permeabilized with saponin (0.05%) and incubated with primary (anti-Akt1/2/3 (Santa Cruz, Dallas, TX, USA)) and oligonucleotide-labeled secondary antibody using standard protocols as for immunofluorescence staining, and TN-RCA performed in situ essentially as described above, washed with PBS, and photographed with a fluorescent microscope (BZ-X710, Keyence, Itasca, IL, USA).
4. Discussion
Trinucleotide rolling circle amplification is a novel isothermal amplification technique that can be used for the sensitive detection and diagnosis of any natural or synthetic DNA or RNA containing short stretches of sequences with only three of the four nucleotides with low background in short time (
Figure 7). Since all the reactions and detection can be performed in liquid form at low temperature, it can be envisioned that TN-RCA can work also on a microfluidics or automated microtiter-based platform, e.g., for large-scale screening of donated blood samples. Similar to RCA, TN-RCA can also be used for in situ detection, semi-quantitative assessment and localization of DNA or RNA (and of specific point mutations) in tissue sections, including frozen and paraffin-embedded tissue sections, in fixed cells, as well as in dried samples, e.g., of dried urine, saliva, blood or in other forensic samples [
9]. For commercial applications, TN-RCA can be envisioned as point of care test (POCT), as clinical, laboratory or field kits or as laboratory technique for in vitro and in situ measurement of specific DNA or RNA. Moreover, TN-RCA can be used to label, detect and identify/authenticate other molecules (e.g., antibodies, proteins, lipids, nucleic acids, organisms/genetically-modified organisms (GMOs), chemicals, solutions such as color in paintings or biometric ink [
55] in writings or microdots [
56]), and other objects of interest that have been tagged or spiked with oligonucleotides (e.g., stabilized with phosphothionate linkage) encoding the complementary target sequence. As reviewed by Ali et al. for RCA [
3], TN-RCA can also be envisioned for numerous applications in nanobiotechnology and materials sciences (e.g., as template for nanoassemblies and nanostructures such as nanowires, nanoribbons and nanotubes, DNA-based metamaterials for drug delivery, hydrogels, DNA glues), with the advantage of lower self-aggregation of the TN-RCA products and lower background due to less formation of multimeric by-products as result of self-annealing of the padlock probes and start primer.
As outlined in the following and exemplified in this study, TN-RCA has several characteristics that may be advantageous over conventional RCA in particular when used at low temperature. First of all, TN-RCA has increased specificity of amplification and lower background. Due to the presence of only three dNTPs (in our case dCTP is missing) in the reaction, only correctly ligated circular G-free padlock probes will amplify and incorporate labeled dNTPs into the TN-RCA product what increases the specific signal and lowers the signals from background amplification from endogenous DNA/RNA. In cases utilizing the presence of a gap between the ends of the padlock, that is either filled in by an enzyme or an oligonucleotide [
7], TN-RCA may have advantages as well since incorrect filled-in ligations that incorporate a G into the padlock probe will not amplify. High molecular weight DNA/RNA and their complexes with proteins that often co-migrate with the RCA product and give background will not be detected (
Figure 2), since fluorescein-12-dUTP is only incorporated after specific amplification into TN-RCA products. Moreover, since all polymerase molecules and dNTPs are used only for specific amplification, they are not consumed in non-specific incorporations, thus enhancing the specific incorporation at lower concentrations. In RCA, background signals can be generated from the presence of high molecular weight genomic DNA in the sample/enzyme preparations [
41], from amplification due to presence of nicks in genomic DNA or RNA (what is the basis of whole-genome amplification [
54]), or from non-specific annealing and/or amplification of padlock probe, start primer, or labelled detection probe to genomic DNA or RNA. The generation of background signals also depends on the method of detection and on the properties and purity of the sample. Labelled dNTPs have been used in conventional RCA [
25] and RCA for in situ detection [
19], but in complex samples they may be incorporated non-specifically giving background, whereas in TN-RCA this does not occur. Optionally, as in RCA, labeled start primers (e.g., with fluorescein, biotin, digoxigenin) can be used for additional labeling, attachment or isolation of the TN-RCA products what may enable also multiplexing and/or automation e.g., using microtiter plates.
A second advantage of TN-RCA is the potentially increased sensitivity and shorter assay time. For detection, when compared using labeled detection probes that usually contain one label per probe and thus also one label per detected concatemeric repeat in the RCA product, TN-RCA can incorporate many and multiple-type labeled dNTP (e.g., the G-free padlock probe used has 22 adenines to incorporate labeled dUTP). Since the labeled dNTPs are incorporated during the TN-RCA, lengthy hybridization procedures can be avoided as they occur for example with microtiter and in situ assays.
A third advantage of TN-RCA is increased specificity at lower reaction temperatures. Due to the absence of guanosine in the G-free padlock probe, or alternatively due to the absence of cytidine in the C-free padlock probe, which are both nucleotides forming Watson–Crick-type triple-bonds with high affinity in DNA and RNA, the overall melting temperature is lower with consequent lower secondary structure of padlock probe and target sequence as well as lower self-priming and self-annealing features of the padlock probe. Similarly, the start primer has a lower secondary structure and self-dimer and self-priming features again reducing background. These features allow to anneal the ends of the padlock probes at lower temperature (e.g., in our case isothermally at 20–37 °C) and/or the usage of longer padlock probe ends for annealing to the target sequences, what leads to a higher specificity and sensitivity. Moreover, since stretches of 20–30 bp or longer target sequences lacking a particular nucleotide are rare in the genome (due to the overall equal presence of all four nucleotides and their random and coding-biased distribution), the specificity of TN-RCA is higher, although it requires the presence of such TN stretches in the target sequences. This requirement can be overcome by the incorporation of “universal base analogues” into the end-sequences of the padlock sequence, in particular when the trinucleotide-target sequence contains one or a few of the forth otherwise missing base to facilitate annealing [
57], but still only three dNTP are required for TN-RCA amplification, thus increasing the number of potential target sequences that are accessible to TN-RCA. The identification of unique TN stretches in natural DNA can be facilitated by developing software able to screen genomic databases such as Genbank [
58].
A disadvantage of TN-RCA is the limit of being able to target only stretches of DNA/RNA containing three or less nucleotides. This may limit the versatility of the method with certain targets (e.g., for shorter genomes of viruses or specific genes). Since after sodium bisulfite treatment of DNA/RNA only cytosine and not 5-methylcytosine is converted to uracil [
59,
60], TN-RCA may also be a method to distinguish un-methylated over methylated cytosines in specific target sequences. Taken together, by targeting only stretches of DNA/RNA containing three or less nucleotides, the TN-RCA method gains features that increase specificity, sensitivity, and lowers background. Similar to the RCA method, variations of the TN-RCA steps and assay conditions can be imagined that increase these features even further.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






