CRISPR-Cas9 Mediated Genome Editing in Bicyclus anynana Butterflies

Abstract
:
1. Introduction
2. Experimental Design
2.1. Experimental Stages
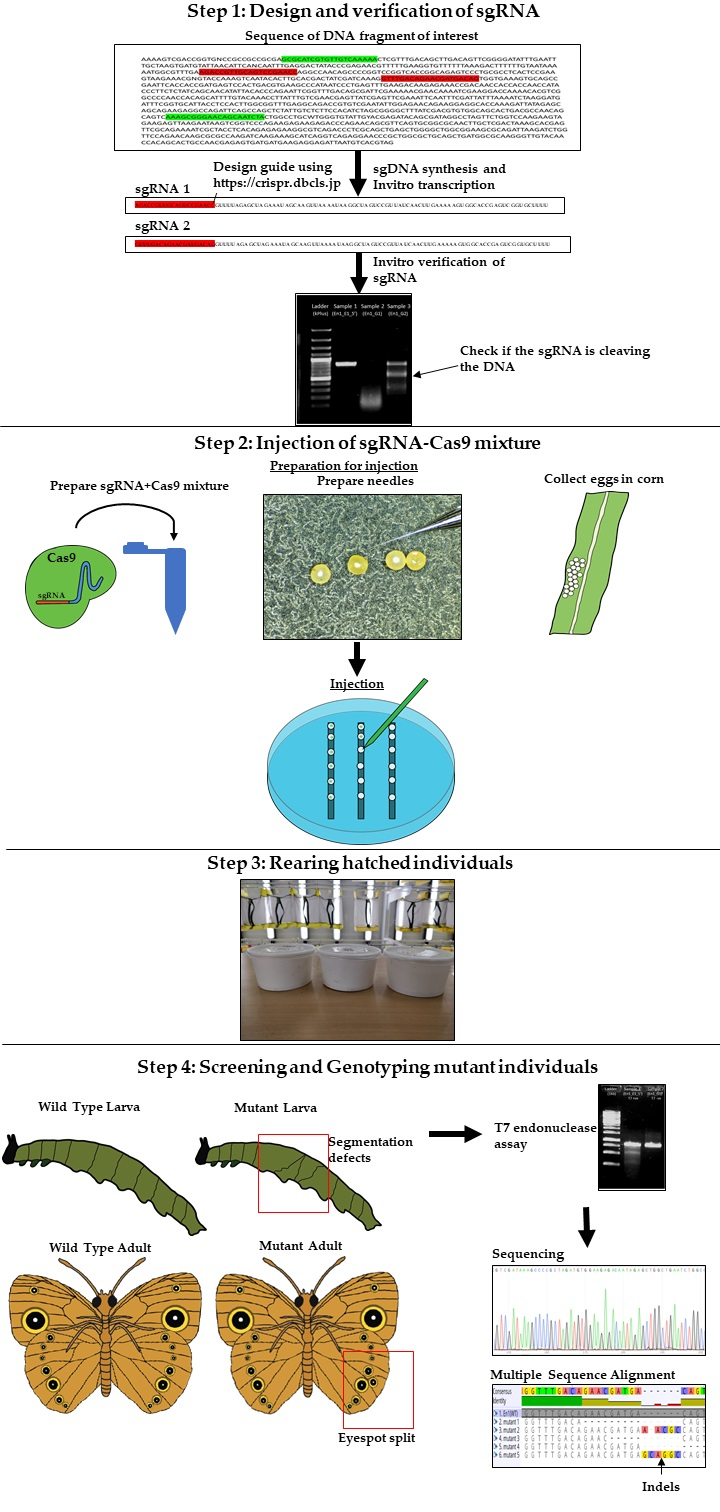
2.1.1. Design, Synthesis and Verification of sgRNA
2.1.2. Injection of the sgRNA-Cas9 Mixture into Embryos
2.1.3. Rearing of Hatched Individuals
2.1.4. Genotyping of Mutant Individuals
2.2. Required Materials and Equipment
2.2.1. Materials
- Microcentrifuge tubes 1.5 mL (Eppendorf, Hamburg, Germany; Cat. no.: T9661-500EA)
- Petri plates (AIT Biotech, Singapore; Cat. no.: 800311)
- Pipette tips—1 mL, 200 µL, 10 µL (Axygen, Union City, CA, USA; Cat. nos.: 14-222-690, 14-222-812, and 14-222-737)
- 20 µL Microloader (Eppendorf; Cat. no.: 5242956003)
- Glass capillary tubes: Glass 1BBL w/FIL 1.0MM 3 IN (World Precision Instruments, Inc., Sarasota, FL, USA; Cat. no.: 1B100F-3)
- PCR tubes 200 µL (Axygen; Cat. no.: 14-222-262)
- 50 mL PYREX Round Bottom glass test tubes (Corning, Corning, NY, USA; Cat. no.: 2619H22)
- 0.5 mm stainless steel beads (Next Advance, Troy, NY, USA; Cat. no.: SKU: SSB05)
- Paper cups and cages (for rearing animals)
2.2.2. Equipment
- Shaking heat block (Thermomixer, Eppendorf)
- Water bath (JULABO TW9; JULABO, Seelbach, Germany)
- Thermocycler (Super Cycler SC300G, Kyratec, Mansfield, Australia)
- Tabletop centrifuge (Eppendorf 5415 D; Eppendorf)
- Refrigerated centrifuge (Eppendorf 5810 R, Eppendorf; Hettich MIKRO 220R, Hettich ZENTRIFUGEN, Frankenberg, Germany)
- Vortex (Genie 2; Scientific Industries, Bohemia, NY, USA)
- −80 °C freezer
- −20 °C freezer
- 4 °C refrigerator
- Needle puller (Flaming/Brown micropipette puller, Model P-97; Sutter Instrument Co., Novato, CA, USA)
- Micro injector (Eppendorf FemtoJet 4i, Eppendorf; Parker Instrumentation Picospritzer III, Parker Instrumentation, Janesville, WI, USA)
- Sanger sequencer (3730xl; Thermo Fisher Scientific, Waltham, MA, USA)
- Pipettes: 200–1000 µL, 20–200 µL, 0.5–10 µL (Eppendorf Research or Research Plus; Eppendorf)
- Gel documentation system (Syngene GeneGenius; Syngene, Frederick, MD, USA)
- Nanodrop ND-1000 Spectrophotometer (Thermo Scientific Nanodrop ND-1000; Thermo Fisher Scientific)
- Microwave (Sharp, Osaka, Japan)
- Weighing balance (Sartorius ENTRIS623i-1S; Sartorius, Göttingen, Germany)
- Imaging system (LEICA DMS 1000; Leica Microsystems, Wetzlar, Germany)
- MilliQ water purification system (Merck Millipore, Burlington, MA, USA)
- Gel electrophoresis system (Bio-Rad, Hercules, CA, USA)
- Vacuum concentrator (Thermo Scientific SAVANT DNA120 SpeedVac Concentrator; Thermo Fisher Scientific)
- Tabletop spinner (Labnet Mini Centrifuge C1201; Labnet, Edison, NJ, USA)
- pH meter (HANNA Edge HI2020-02; HANNA, Woonsocket, RI, USA)
- Class II Biological Safety cabinet (LabCard, Lenexa, KS, USA)
- Homogenizer (Next Advance Bullet Blender; Next Advance)
- Autoclave (HIRAYAMA, Saitama, Japan)
2.2.3. Reagents for sgRNA Synthesis
- Ultramers 100 mM (Sigma-Aldrich, St. Louis, MO, USA; En1_G1, En1_G2 and CRISPR_R; see Table 2 for sequence
- dNTP mix (Promega, Madison, WI, USA; Cat. no.: U1511; NEB, Ipswich, MA, USA; Cat. no.: N0447S)
- Q5 high fidelity DNA polymerase (NEB; Cat. no.: M0491S)
- Q5 polymerase buffer 10X (NEB; Cat. no.: B9027S)
- Molecular grade water (HyPure Molecular Biology Grade Water, HyClone, Thermo Fisher Scientific; Cat. no.: 7732-18-5)
- Agarose molecular grade (Vivantis, Selangor Darul Ehsan, Malaysia; Cat. no.: PC0701-500g)
- Trizma base (Sigma-Aldrich; Cat. no.: T1503-500G)
- EDTA (ThermoFisher Scientific; Cat. no.: 17892)
- Acetic acid (Sigma-Aldrich; Cat. no.: 64-19-7)
- SYBRSafe (Invitrogen, Carlsbad, CA, USA; Cat. no.: S33102)
- GeneJet PCR purification kit (Thermo Fisher Scientific; Cat. no.: K0702)
- T7 10× buffer (NEB; Cat. no.: M0251S)
- T7 RNA polymerase (NEB; Cat. no.: M0251S)
- ATP, GTP, CTP and UTP: 5 µmol (Thermo Fisher Scientific; Cat. nos.: 18330-019, 18331-017, 18332-015, and 18333-013)
- Ribolock RNase inhibitor (Thermo Fisher Scientific; Cat. no.: EO0381)
- DNA ladder (GeneDireX Kplus, Keelung City, Taiwan, Cat. No: DM011-R500/; Invitrogen 1 Kb and KbPlus DNA ladder, Thermo Fisher Scientific; Cat. nos.: 10787018 and SM0313)
- DNase I (Thermo Fisher Scientific; Cat. no.: EN0525)
- 10× DNase I buffer (Thermo Fisher Scientific; Cat. no.: AM8170G)
- NaOAc (Sigma-Aldrich; Cat. no.: S2889)
- Ethanol 100% (VWR chemicals, VWR, Radnor, PA, USA; Cat. no.: 64-17-5)
- 6× DNA loading dye (Thermo Fisher Scientific; Cat. no.: R0611)
- Riboruler High Range RNA Ladder 20 (Thermo Fisher Scientific; Cat. no.: SM1821)
- 2× RNA loading Dye (Thermo Fisher Scientific; Cat. no.: R0641)
2.2.4. Reagents for sgRNA In Vitro Verification
- Tissue DNA extraction Kit (E.Z.N.A Tissue DNA Kit, Omega Bio-tek, Norcross, GA, USA; Cat. no.: D3396-02)
- 2× PCRBIO Taq Mix Red (PCR Biosystems, London, UK; Cat. no.: PB10.11-20)
- Oligonucleotides 100 mM (Integrated DNA Technologies, Coralville, IA, USA; En1_E1_5′_F and En1_E1_5′_R; see Table 24 and Figure 2 for sequence)
- Molecular grade water (HyPure Molecular Biology Grade Water, HyClone, Thermo Fisher Scientific; Cat. no.: 7732-18-5)
- Cas9 Protein NLS (NEB; Cat. no.: M0641)
- Cas9 buffer 10× (NEB; Cat. no.: M0641)
- Agarose Molecular grade (Vivantis; Cat. no.: PC0701-500g)
- SYBRSafe (Invitrogen; Cat. no.: S33102)
- PCR purification kit (Thermo Fisher Scientific GeneJET PCR Purification Kit; Cat. no.: K0701)
2.2.5. Reagents for CRISPR Injection
- Cas9 Protein NLS (NEB; Cat. no.: M0641)
- Cas9 buffer 10× (NEB; Cat. no.: M0641)
- Non-toxic food dye (Star Brand Artificial True Blue Color, Star Brand)
- Molecular grade water (HyPure Molecular Biology Grade Water, HyClone, Thermo Fisher Scientific; Cat. no.: 7732-18-5)
2.2.6. Reagents for Isolation of Genomic DNA from Injected Embryos/Larvae/Adults
2.2.7. Reagents for Cloning
- Competent cells (prepared in-house; for protocol see ref. [58])
- pGEM-T Vector System (Promega; Cat. no.: A3600)
- Luria–Bertani (LB) agar (Invitrogen; Cat. no.: 22700025)
- MilliQ water (Merck Millipore)
- Ampicillin (Sigma-Aldrich; Cat. no.: 10835242001)
- Isopropyl β-d-1-thiogalactopyranoside (IPTG; Ambion, Foster City, CA, USA; Cat. no.: AM9464)
- X-Gal (Ambion; Cat. no.: 15520034)
- LB base (Invitrogen; Cat. no.: 12780029)
2.2.8. Reagents for Genotyping
- Plasmid isolation kit (GeneJET Plasmid Miniprep Kit, Thermo Fisher Scientific; Cat. no.: K0502)
- Oligonucleotide (Integrated DNA Technologies; M13F and M13R; for sequence see Table 3)
- BigDye Terminator v3.1 RR-5000 and sequencing buffer (Thermo Fisher Scientific; Cat. no.: 4337457)
- PCR purification kit (Thermo Fisher Scientific GeneJET PCR Purification Kit; Cat. no.: K0701)
3. Procedure
3.1. Design, Synthesis, and Purification of sgRNA. Time for Completion: 3 Days
3.1.1. Design of sgRNA
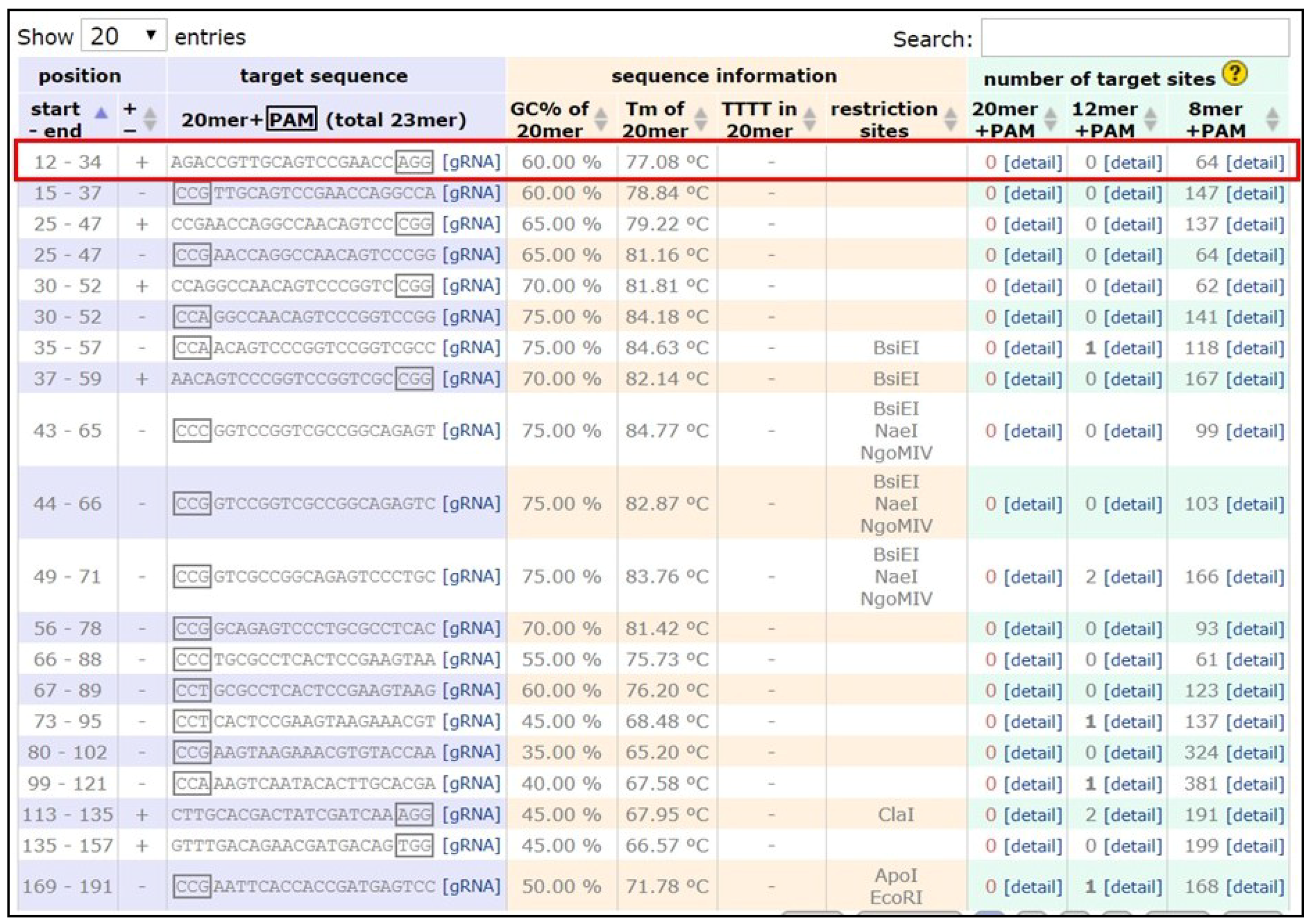
- Identify the DNA sequence to be modified. Two versions of the B. anynana genome are available on lepbase [60]. Genes can be searched using the search field.
- Copy the DNA sequence and go to the webpage mentioned in [59]. Paste the sequence in the ‘Paste a nucleotide sequence’ box.
- Apply the filter ‘Postman butterfly (Heliconius melpomene) genome, Hmel1 (February 2012)’ or ‘Monarch butterfly (Danaus plexippus) genome, DanPle_1.0 (November 2011)’ in the specificity checkbox and click the design bar.
- Copy a candidate target sequence (see Figure 3) with orientation 5′ to 3′ (+) without the PAM sequence (Note: sequences closer to the 5′ region of the gene of interest and having high GC content should be preferred) and insert it in the grey highlighted part of the template below:5′-GAAATTAATACGACTCACTATAGG-xxxxxxxxxxxxxxxxxxxxx-GTTTTAGAGCTAGAAATAGC-3′This is the forward primer for guide synthesis.The reverse primer for sgRNA synthesis is the same for all target sites (see below):5′-AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3′.
3.1.2. Synthesis of sgDNA
- Add molecular grade water to the lyophilized primers in their original tubes (forward and reverse) to make a stock solution of 100 mM. Prepare a working solution of 10 mM by combining 10 µL of the stock solution with 90 µL molecular grade water in a new set of 1.5 mL tubes.
- Add reagents as mentioned in Table 4 in a 200 µL PCR tube:
![Mps 01 00016 i001]() CRITICAL STEP: Use high fidelity DNA polymerase only. Prepare at least three tubes for each sgDNA to obtain enough yield.
CRITICAL STEP: Use high fidelity DNA polymerase only. Prepare at least three tubes for each sgDNA to obtain enough yield. - Setup the PCR reaction with the conditions mentioned in Table 5:
- Run the PCR reaction in 1% agarose gel.
![Mps 01 00016 i002]() PAUSE STEP: The PCR reaction mixture can be stored at 4 °C overnight.
PAUSE STEP: The PCR reaction mixture can be stored at 4 °C overnight.


3.1.3. Purification of sgDNA (Using Thermo Scientific GeneJET PCR Purification Kit)
- Transfer the completed reaction volume to a 1.5 mL microcentrifuge tube and add an equal volume of binding buffer. Vortex the mixture for 5 s.
- Transfer the mixture to the GeneJET PCR purification column and centrifuge at 13,000 rpm for 30 s. Discard the flow through.
- Add 500 µL of wash buffer and centrifuge at 13,000 rpm for 30 s. Discard the flow through and repeat this step one more time.
- Spin the moist column for one additional min at 13,000 rpm and discard the collection tube.
- Transfer the column to a new 1.5 mL microcentrifuge tube and add 20 µL of elution buffer or molecular grade water. Incubate the column at room temperature for 3–5 min.
- Centrifuge at 13,000 rpm for 1 min and measure the concentration of the elution using Nanodrop.
![Mps 01 00016 i002]() PAUSE STEP: Prepare a working concentration of 500 ng/µL. The purified DNA can be stored at 4 °C for over one month. For long-term storage, use a −20 °C freezer.
PAUSE STEP: Prepare a working concentration of 500 ng/µL. The purified DNA can be stored at 4 °C for over one month. For long-term storage, use a −20 °C freezer.
3.1.4. In Vitro Transcription to Prepare sgRNA
- Add the reagents mentioned in Table 6 in a 1.5 mL microcentrifuge tube:
- Incubate the mixture in water bath at 37 °C for 16 h.
- Add 1 µL of DNaseI and 2 µL of 10× DNaseI buffer.
- Incubate the mixture in water bath at 37 °C for 15 min.
- Remove 1 µL of the reaction mixture in a 200 µL PCR tube. Add 7 µL of molecular grade water and 2 µL of 2× RNA loading dye.
- Heat the sample at 70 °C for 10 min and run it in 1% agarose gel.
![Mps 01 00016 i001]() CRITICAL STEP: RNA degrades very fast. To prevent degradation properly, clean the gel tray using MilliQ water, use fresh buffer and run the gel for 15–20 min at low voltage.
CRITICAL STEP: RNA degrades very fast. To prevent degradation properly, clean the gel tray using MilliQ water, use fresh buffer and run the gel for 15–20 min at low voltage.
3.1.5. Purification of sgRNA (via Ethanol Precipitation)
- Add 80 µL of molecular grade water to the reaction tube from the previous step to raise the volume to 100 µL.
- Add 10 µL of 3 M NaOAc and 200 µL of 100% ethanol.
- Vortex the mixture for 10 s and store at −20 °C for 15–20 min.
- Centrifuge the mixture at 4 °C, 14,000 rpm for 15 min.
- Carefully remove the supernatant.
![Mps 01 00016 i001]() CRITICAL STEP: Be very careful not to disturb the pellet.
CRITICAL STEP: Be very careful not to disturb the pellet. - Dry the sample in a vacuum concentrator and add 20 µL of molecular grade water.
- Prepare a stock concentration of 600 ng/µL by adding additional water (after a Nanodrop reading) and store aliquots at −20 °C.
![Mps 01 00016 i002]() PAUSE STEP: RNA can be stored at −20 °C for over 1 year.
PAUSE STEP: RNA can be stored at −20 °C for over 1 year.
3.2. OPTIONAL STEP: Preparation and Purification of Cas9 mRNA. Time for Completion: 2 Days
3.2.1. Preparation of Cas9 mRNA (Using mMESSAGE mMACHINE T3 Kit and Poly(A) Tailing Kit, ThermoFisher Scientific)
- Add 10 µg of Addgene plasmid #46757 (pT3TS-nCas9n), 2 µL of restriction enzyme and molecular grade water (to make up the volume to 20 µL) in a 1.5 mL microcentrifuge tube.
- Incubate the mixture at 37 °C for 2 h.
- Add the reagents mentioned in Table 7 in a 1.5 mL centrifuge tube:
- Incubate the mixture in water bath at 37 °C for 4 h.
- Add 1 µL of DNase I and 2 µL of 10× DNase buffer and incubate in water bath at 37 °C for 15 min.
- For poly(A) tailing add the reagents mentioned in Table 8 to the tube above:
- Incubate the mixture in water bath at 37 °C for 1 h.
3.2.2. Purification of Cas9 mRNA
- Add 60 µL of LiCl2 (provided in the mMESSAGE mMACHINE kit) to the tube above and incubate at −20 °C for 30 min.
- Centrifuge at 4 °C, 14,000 rpm for 15 min.
- Carefully remove the supernatant and resuspend the pellet in 100 µL 70% ethanol.
- Centrifuge at 4 °C, 14,000 rpm for 15 min.
- Remove the supernatant and dry the sample in a vacuum concentrator.
- Resuspend the pellet in 10 µL molecular grade water and measure the concentration using Nanodrop. Store the RNA at −20 °C.
![Mps 01 00016 i002]() PAUSE STEP: RNA can be stored at −20 °C for over one year.
PAUSE STEP: RNA can be stored at −20 °C for over one year.
3.3. Isolation of the Fragment of Interest (e.g., from Genomic DNA, Plasmid, etc.) and Verification of sgRNA via Invitro Cleavage. Time for Completion: 1 Day
3.3.1. Isolation of Genomic DNA (Using E.Z.N.A Tissue DNA Kit, Omega Bio-tek)
- Remove the epidermis of 5th instar larvae and transfer the tissue into a 1.5 mL microcentrifuge tube.
![Mps 01 00016 i001]() CRITICAL STEP: Carefully remove the gut material from the larvae as it might contaminate the DNA sample. One larva should be enough for a yield of around 500 ng/µL in 100 µL volume. Alternatively, tissues can be extracted from the thorax of adult Bicyclus or embryos.
CRITICAL STEP: Carefully remove the gut material from the larvae as it might contaminate the DNA sample. One larva should be enough for a yield of around 500 ng/µL in 100 µL volume. Alternatively, tissues can be extracted from the thorax of adult Bicyclus or embryos. - Add 200 µL TL buffer and homogenize the tissue in homogenizer using 0.5 mm stainless steel beads for 5 min.
- Add 20 µL OB protease solution and vortex for 10 s.
- Incubate the mixture in shaking heat block at 55 °C for 16 h.
- Centrifuge the tube at 14,000 rpm for 5 min to precipitate the cell debris.
- Transfer the supernatant to a fresh 1.5 mL microcentrifuge tube and add 220 µL BL buffer.
- Incubate the mixture in water bath at 70 °C.
- Add 220 µL 100% ethanol and vortex for 10 s.
- Transfer the mixture to HiBind DNA column and centrifuge at 14,000 rpm for 1 min.
- Discard the filtrate and add 500 µL HBC buffer.
- Centrifuge at 14,000 rpm for 30 s and discard the filtrate.
- Transfer the column to a fresh 2.0 mL collection tube.
- Add 500 µL DNA wash buffer and centrifuge at 14,000 rpm for 30 s. Discard the flow through and repeat this step one more time.
- Centrifuge the empty column at 14,000 rpm for 1 min and transfer the column into a fresh 1.5 mL microcentrifuge tube.
- Add 100 µL elution buffer or molecular grade water to the column and let it sit for 5 min at room temperature.
- Centrifuge at 14,000 rpm for 1 min and measure the concentration using Nanodrop. Store the DNA at 4 °C for immediate use.
![Mps 01 00016 i002]() PAUSE STEP: DNA can be stored at −20 °C for over three years.
PAUSE STEP: DNA can be stored at −20 °C for over three years.
3.3.2. Design of Primers for Amplification of DNA Fragment of Interest.
- Copy the sequence of interest and paste it into the box on the webpage mentioned in ref [61].
- Under the ‘General Setting’ tab change the values:Primer Tm: Min: 55; Max: 65Primer GC%: Min: 45; Opt: 60; Max: 60Max Tm Difference: 3
- Click on the ‘Pick Primers’ tab in the top right corner.
- Select the best set from the list of primers.
3.3.3. Amplification of DNA Fragment of Interest
- Resuspend the lyophilized primers using molecular grade water to make a stock solution of 100 ng/µL. Prepare a working solution of 10 mM as described above.
- Add the reagents mentioned in Table 9 in a 200 µL PCR tube:
![Mps 01 00016 i001]() CRITICAL STEP: Prepare at least five tubes in order to identity the most optimal annealing temperature in a gradient PCR reaction.
CRITICAL STEP: Prepare at least five tubes in order to identity the most optimal annealing temperature in a gradient PCR reaction. - Setup the gradient PCR reaction with conditions as mentioned in Table 10:
- Run the reaction mixture in 1% agarose gel for 30 min.
![Mps 01 00016 i002]() PAUSE STEP: The PCR reaction mixture can be stored at 4 °C overnight.
PAUSE STEP: The PCR reaction mixture can be stored at 4 °C overnight.
3.3.4. Purification of Amplified DNA (Using ThermoFisher Scientific GeneJET PCR Purification Kit)
- Transfer the reaction volume to a 1.5 mL microcentrifuge tube and add an equal volume of binding buffer. Vortex the mixture for 5 s.
- Transfer the mixture to the GeneJET PCR purification column and centrifuge at 13,000 rpm for 30 s. Discard the flow through.
- Add 500 µL of wash buffer and centrifuge at 13,000 rpm for 30 s. Discard the flow through and repeat this step one more time.
- Spin the moist column for and additional min at 13,000 rpm and discard the collection tube.
- Transfer the column to a new 1.5 mL microcentrifuge tube and add 20 µL of elution buffer or molecular grade water. Incubate the column at room temperature for 3–5 min.
- Centrifuge at 13,000 rpm for 1 min and measure the concentration using Nanodrop.
![Mps 01 00016 i002]() PAUSE STEP: The purified DNA can be stored at 4 °C for over one month. For long-term storage use a −20 °C freezer.
PAUSE STEP: The purified DNA can be stored at 4 °C for over one month. For long-term storage use a −20 °C freezer.
3.3.5. Verification of sgRNA via In Vitro Cleavage
- Add the reagents mentioned in Table 11 in a 1.5 mL microcentrifuge tube:
- Incubate the sample in a water bath at 37 °C for 10 min.
- Add 200 ng of the amplified DNA of interest with the recognition site and incubate the mixture in the water bath at 37 °C for 20 min.
- Run 5 µL of the mixture along with 100 ng of the original template DNA in 1% agarose gel for 30 min.
3.3.6. OPTIONAL STEP: Alternative Verification of sgRNA (If Step 3.3.5 Does Not Work)
- Add the reagents mentioned in Table 12 in a 1.5 mL microcentrifuge tube:
- Add 0.1 µL of Cas9 Protein NLS (3300 ng/µL) and incubate the mixture at 25 °C for 10 min.
- Add 100 µg/mL BSA (Bovine Serum Albumin) and incubate the mixture in a water bath at 37 °C for 3 h.
- Add 1 µL of proteinase K and incubate the mixture in a water bath at 37 °C for 10 min.
3.4. Embryo Collection, Needle Preparation, and Injection of the sgRNA-Cas9 Mixture: Time for Completion: 1 Day
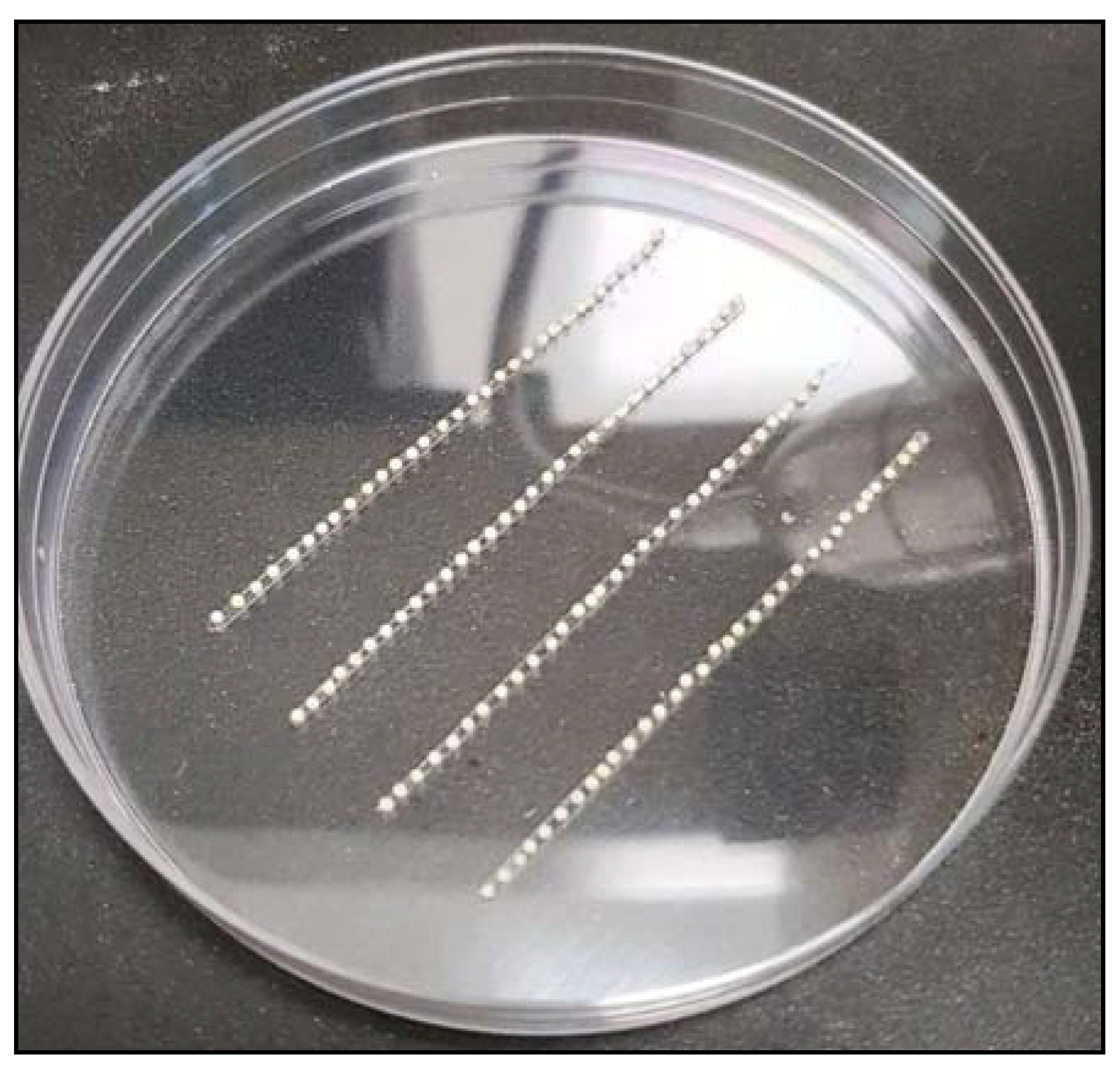
3.4.1. Needle Preparation
- Turn on the needle puller machine and set a program with the settings as mentioned in Table 13:
- Arrest the glass capillary in place and click on the enter button. Wait for the process of heating and pulling the glass to be completed.
- Carefully remove the needles from the machine and place them on plasticine (as shown in Figure 4).
3.4.2. Embryo Collection
- Place a few young corn leaves inside butterfly cages and leave them for 30 min. (Note: The best time for embryo collection is around 2:00 p.m. to 3:00 p.m.).
- Remove the leaves and collect the embryos in a paper cup.
- Prepare a Petri dish with thin strips of double-sided tape attached to the bottom of the plate.
- Using a paintbrush, carefully arrange the embryos on the double-sided tape (Figure 5).
3.4.3. Preparation of sgRNA-Cas9 Mixture
- Add the reagents mentioned in Table 14 in a 1.5 mL microcentrifuge tube:
- Incubate the mixture in a water bath at 37 °C for 10 min.
- Add 0.5 µL non-toxic food dye and store at room temperature until use.
3.4.4. Injection of the sgRNA-Cas9 Mixture into Embryos
- Turn on the injector and set the conditions as follows:For PICOSPRITZER IIIDURATION: 30 millisecondsFor FemtoJet 4ipi[PSI] = 0.50; pc[PSI] = 0.10 for t[sec] = 0.50
- Pipette 3 µL of the sgRNA-Cas9 mix using a 20 µL Microloader tip and transfer the content to the needle by filling it from the back.
- Attach the needle to the injection holder and break the tip of the needle (e.g., remove the molten glass at the tip that obstructs the opening) by gently pressing against the side of a Petri dish under a dissecting microscope.
![Mps 01 00016 i001]() CRITICAL STEP: Be careful while breaking the needle tip. If the desired sharpness is not attained, transfer the mixture back to the 1.5 mL tube (by pushing it out with air pressure) and repeat the steps above.
CRITICAL STEP: Be careful while breaking the needle tip. If the desired sharpness is not attained, transfer the mixture back to the 1.5 mL tube (by pushing it out with air pressure) and repeat the steps above. - Inject the embryos under a dissecting microscope until the mixture is visible inside the embryos.
3.5. Rearing Hatchling and Screening for Mutants. Time for Completion: 4–5 Weeks
Rearing of the Hatchlings (Condition for Rearing Include 12-12 Day-to-Night Cycle, Temperature of 27 °C, and Humidity of 80%).
- Place a wet cotton ball inside the Petri plate above and incubate at 27 °C. It will take 3–4 days for the embryos to hatch (use the unhatched embryos for the T7 endonuclease assay).
- Transfer the hatched larvae into a paper cup with one young corn leaf. Keep feeding these larvae inside cups with fresh cut leaves until they reach the 3rd instar. Transfer these older larvae to larger rearing cages. Keep note of any changes in phenotype and image these abnormal individuals/tissues.
- Pupae are assigned to separate paper or plastic cups, individually, where adults emerge after one week.
- Freeze the adults at −20 °C for imaging and genotyping.
3.6. T7 Endonuclease Assay and Genotyping of Mutants. Time for Completion: 3 Days
3.6.1. Isolation of Genomic DNA from Mutant Tissue (using E.Z.N.A Tissue DNA Kit)
- Remove mutant clones of cells (such as epidermis of larvae with segmentation defect, wing region with extra eyespot etc.) and transfer into a 1.5 mL microcentrifuge tube.
![Mps 01 00016 i001]() CRITICAL STEP: Carefully remove the gut material from the larvae to minimize contaminating the larval DNA with bacterial DNA.
CRITICAL STEP: Carefully remove the gut material from the larvae to minimize contaminating the larval DNA with bacterial DNA. - Add 200 µL TL buffer and homogenize the tissue in homogenizer using 0.5 mm stainless steel beads for 5 min.
- Add 20 µL OB protease solution and vortex for 10 s.
- Incubate the mixture in a shaking heat block at 55 °C for 16 h.
- Centrifuge the tube at 14,000 rpm for 5 min to precipitate the cell debris.
- Transfer the supernatant to a fresh 1.5 mL microcentrifuge tube and add 220 µL BL buffer.
- Incubate the mixture in a water bath at 70 °C.
- Add 220 µL of 100% ethanol and vortex for 10 s.
- Transfer the mixture to a HiBind DNA column and centrifuge at 14,000 rpm for 1 min.
- Discard the filtrate and add 500 µL of HBC buffer.
- Centrifuge at 14,000 rpm for 30 s and discard the filtrate.
- Transfer the column to a fresh 2.0 mL collection tube.
- Add 500 µL DNA wash buffer and centrifuge at 14,000 rpm for 30 s. Discard the flow through and repeat this step one more time.
- Centrifuge the empty column at 14,000 rpm for 1 min and transfer the column into a fresh 1.5 mL microcentrifuge tube.
- Add 100 µL elution buffer or molecular grade water to the column and let it sit for 5 min at room temperature.
- Centrifuge at 14,000 rpm for 1 min and measure the concentration using Nanodrop. Store the DNA at 4 °C for immediate use.
![Mps 01 00016 i002]() PAUSE STEP: DNA can be stored at −20 °C for over three years.
PAUSE STEP: DNA can be stored at −20 °C for over three years.
3.6.2. Amplification of DNA Fragment of Interest from DNA Isolated from Mutant Tissue
- Add the reagents mentioned in Table 15 in a 200 µL PCR tube:
![Mps 01 00016 i001]() CRITICAL STEP: Prepare at least five tubes to identify the most optimal annealing temperature using gradient PCR.
CRITICAL STEP: Prepare at least five tubes to identify the most optimal annealing temperature using gradient PCR. - Setup the PCR reaction with the conditions mentioned in Table 16:
- Run the reaction mixture in 1% agarose gel for 30 min.
![Mps 01 00016 i002]() PAUSE STEP: The PCR reaction mixture can be stored at 4 °C overnight.
PAUSE STEP: The PCR reaction mixture can be stored at 4 °C overnight.
3.6.3. Purification of Amplified DNA (Using ThermoFisher Scientific GeneJET PCR Purification Kit)
- Transfer the completed reaction volume to a 1.5 mL microcentrifuge tube and add an equal volume of binding buffer. Vortex the mixture for 5 s.
- Transfer the mixture to the GeneJET PCR purification column and centrifuge at 13,000 rpm for 30 s. Discard the flow through.
- Add 500 µL of wash buffer and centrifuge at 13,000 rpm for 30 s. Discard the flow through and repeat this step one more time.
- Centrifuge the empty column for one additional min at 13,000 rpm and discard the collection tube.
- Transfer the column to a fresh 1.5 mL microcentrifuge tube and add 20 µL of elution buffer or molecular grade water. Incubate the column at room temperature for 3–5 min.
- Centrifuge at 13,000 rpm for 1 min and measure the concentration using Nanodrop.
![Mps 01 00016 i002]() PAUSE STEP: Prepare a working concentration of 200 ng/µL. The purified DNA can be stored at 4 °C for over one month. For long-term storage use a −20 °C freezer.
PAUSE STEP: Prepare a working concentration of 200 ng/µL. The purified DNA can be stored at 4 °C for over one month. For long-term storage use a −20 °C freezer.
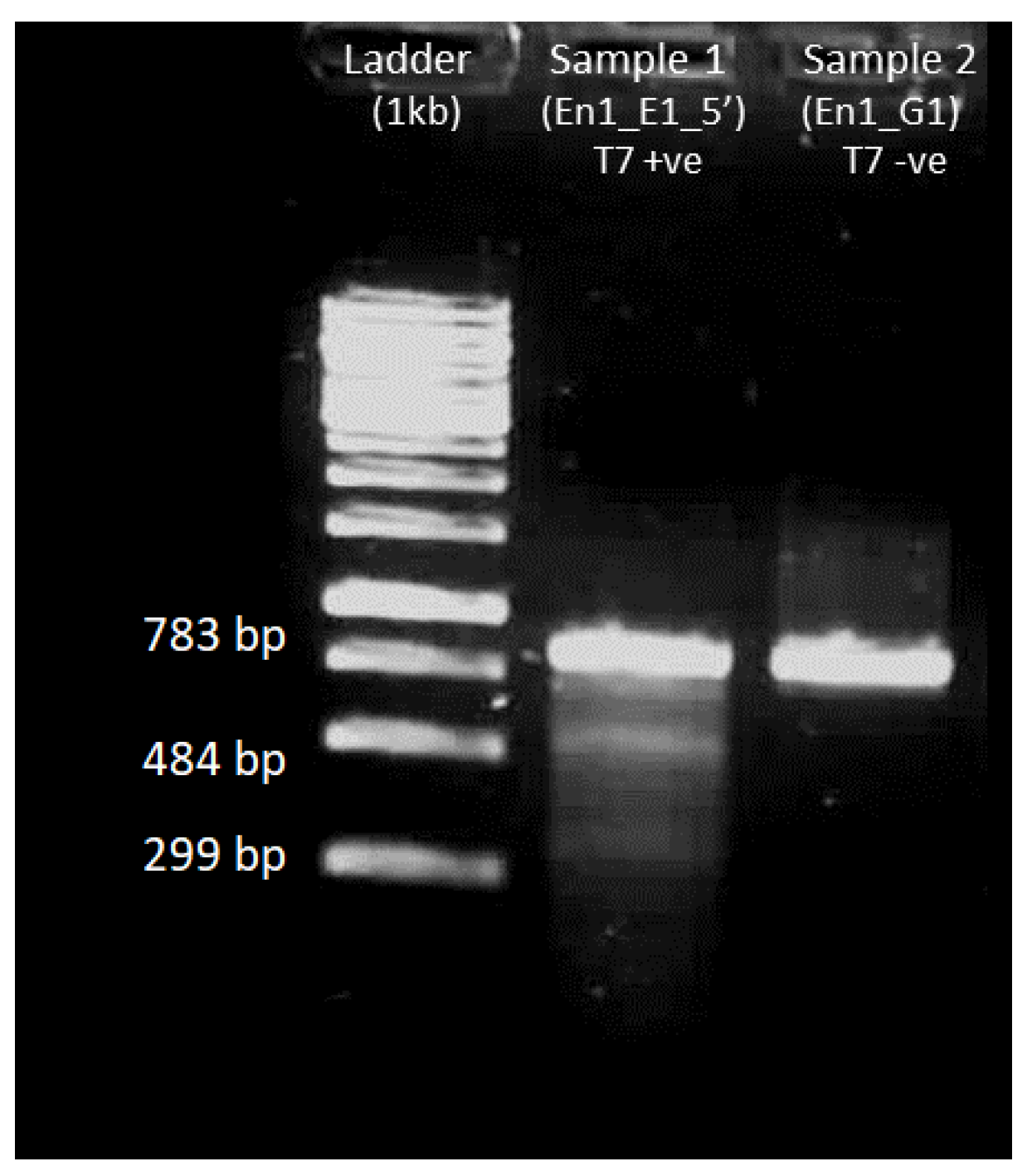
3.6.4. T7 Endonuclease Assay on Amplified DNA Fragments
- Prepare two 200 µL PCR tubes and add the reagents mentioned in Table 17 into each of them:
- Perform T7 hybridization in thermocycler with the conditions mentioned in Table 18:
- Add 1 µL T7 endonuclease in one tube and incubate both tubes in water at 37 °C for 15 min.
- Run the samples in 1% agarose gel for 30 min.
3.6.5. Cloning of amplified DNA fragments (using pGEM-T Vector System)
- Add the reagents mentioned in Table 19 in a 1.5 mL microcentrifuge tube (ligation mixture):
- Incubate the reaction mixture at 4 °C for 16 h.
- Take out one vial of competent cells and keep the tube on ice for 15 min.
- Transfer 5 µL of ligation mixture into the competent cell tube and tap gently to mix the solution.
- Leave the mixture on ice for 30 min.
- Heat shock the cells by transferring the tube into a water bath at 42 °C for 45 s.
![Mps 01 00016 i001]() CRITICAL STEP: Be careful not to exceed the heat shock step above 45 s.
CRITICAL STEP: Be careful not to exceed the heat shock step above 45 s. - Transfer the tube into ice and leave it for 2 min.
- Add 500 µL of autoclaved LB broth and incubate the cells in bacterial incubation chamber at 37 °C with shaking speed of 225 rpm for 2 h.
- Centrifuge the tube at 3000 rpm for 4 min.
- Inside a biological safety cabinet add the reagents mentioned in Table 20 to an LB agar plate:
- Spread the reagents on the plate using glass beads and let the plate dry inside the hood.
- Add 50 µL of supernatant from step 9 and spread across the plate using the glass beads.
- Once dried, seal the plate using parafilm and incubate the plate inside a bacterial incubator at 37 °C for 14 h.
3.6.6. Colony PCR on Transformed Clones
- In a 1.5 mL microcentrifuge tube, add 10 µL molecular grade water. Pick a transformed white colony and transfer it into the tube. Vortex gently to homogenize the colony.
![Mps 01 00016 i001]() CRITICAL STEP: Prepare at least 10 clones (colonies) for testing.
CRITICAL STEP: Prepare at least 10 clones (colonies) for testing. - Add the reagents mentioned in Table 21 in 200 µL PCR tubes:
- Setup the PCR reaction with the conditions mentioned in Table 22:
- Run the reaction mixture in a 1% agarose gel for 30 min and note down the colonies with a single band of the expected size, e.g., those that don’t have an empty plasmid.
- Inside a laminar hood, add 5 µL of ampicillin stock solution into a test tube with 5 mL LB broth. Transfer 5 µLs of homogenized cells from step 1. Do this step for every positive colony.
- Incubate the tubes in a bacterial incubation chamber at 37 °C and 225 rpm for 14–16 h.
3.6.7. Isolation of Plasmids from Transformed Clones (Using GeneJET Plasmid Miniprep Kit)
- Harvest the cells in a 1.5 mL centrifuge tube at 3000 rpm for 5 min (pellet can be stored in 40% glycerol at −80 °C for future use).
- Discard the supernatant and resuspend the pellet in 250 µL of resuspension buffer.
- Add 250 µL of lysis buffer and mix by inverting the tube 6–10 times.
- Add 350 µL of neutralization buffer and mix by inverting the tube 6–10 times.
- Centrifuge at 14,000 rpm for 5 min and transfer the supernatant to GeneJET spin column.
- Centrifuge the column at 14,000 rpm for 30 s.
- Add 500 µL of wash buffer and centrifuge at 14,000 rpm for 30 s. Discard the flow through and repeat this step one more time.
- Centrifuge the empty column at 14,000 rpm for 1 min.
- Transfer the column to a 1.5 mL microcentrifuge tube and add 20 µL of elution buffer or molecular grade water. Incubate the mixture at room temperature for 3 min.
- Centrifuge the column at 14,000 rpm for 1 min and measure the concentration of plasmid using Nanodrop.
![Mps 01 00016 i002]() PAUSE STEP: Prepare a working concentration of 100 ng/µL. The purified plasmid can be stored at 4 °C for over one month. For long-term storage, use a −20 °C freezer.
PAUSE STEP: Prepare a working concentration of 100 ng/µL. The purified plasmid can be stored at 4 °C for over one month. For long-term storage, use a −20 °C freezer.
3.6.8. Sequencing of Cloned DNA Fragments
- Add the reagents mentioned in Table 23 in a 200 µL PCR tube:Add the reagents mentioned in Table 24 in another 200 µL PCR tube:
- Setup sequencing PCR reaction with the conditions mentioned in Table 25:
- In 1.5 mL microcentrifuge tubes, add 40 µL molecular grade water and transfer the reaction mix from the previous step.
- Vortex the mix and incubate at −20 °C for 20 min.
- Centrifuge at 4 °C, 14,000 rpm for 15 min.
- Remove the supernatant and resuspend the pellet in 100 µL 70% ethanol.
- Centrifuge at 4 °C, 14,000 rpm for 15 min.
- Carefully remove the supernatant and dry the sample in a vacuum concentrator.
- Store at −20 °C until sequencing.
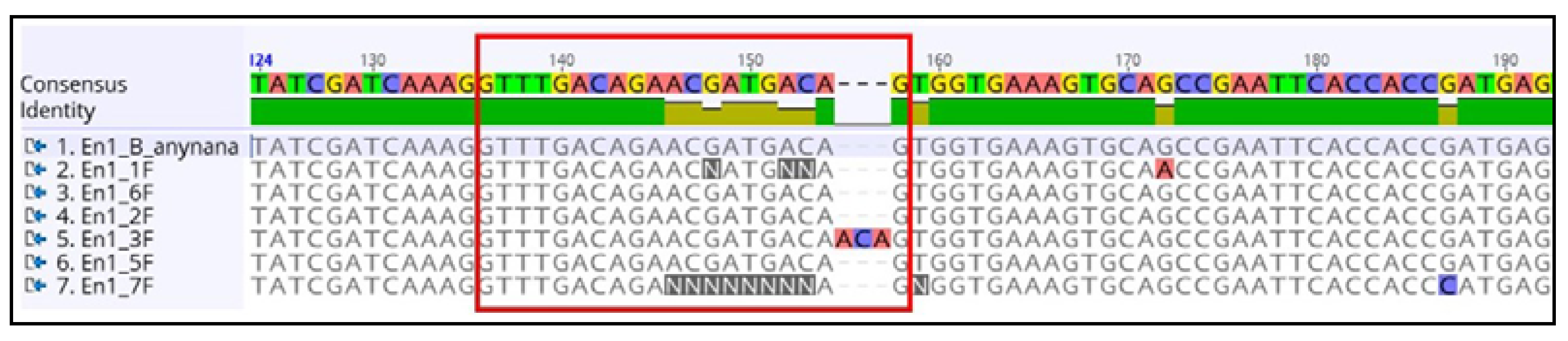
3.6.9. Analyzing the Sequencing Results and Determination of Indel Sites
- Copy the sequences in a word file with specific identifiers.
- Align the sequences along with the original DNA sequence using ClustalW [63] or the Geneious multiple sequence alignment tool.
- Look for the indels at the expected site and save the file for future use.
4. Expected Results
4.1. Design, Synthesis and Purification of sgRNA
4.1.1. Yield of sgDNA
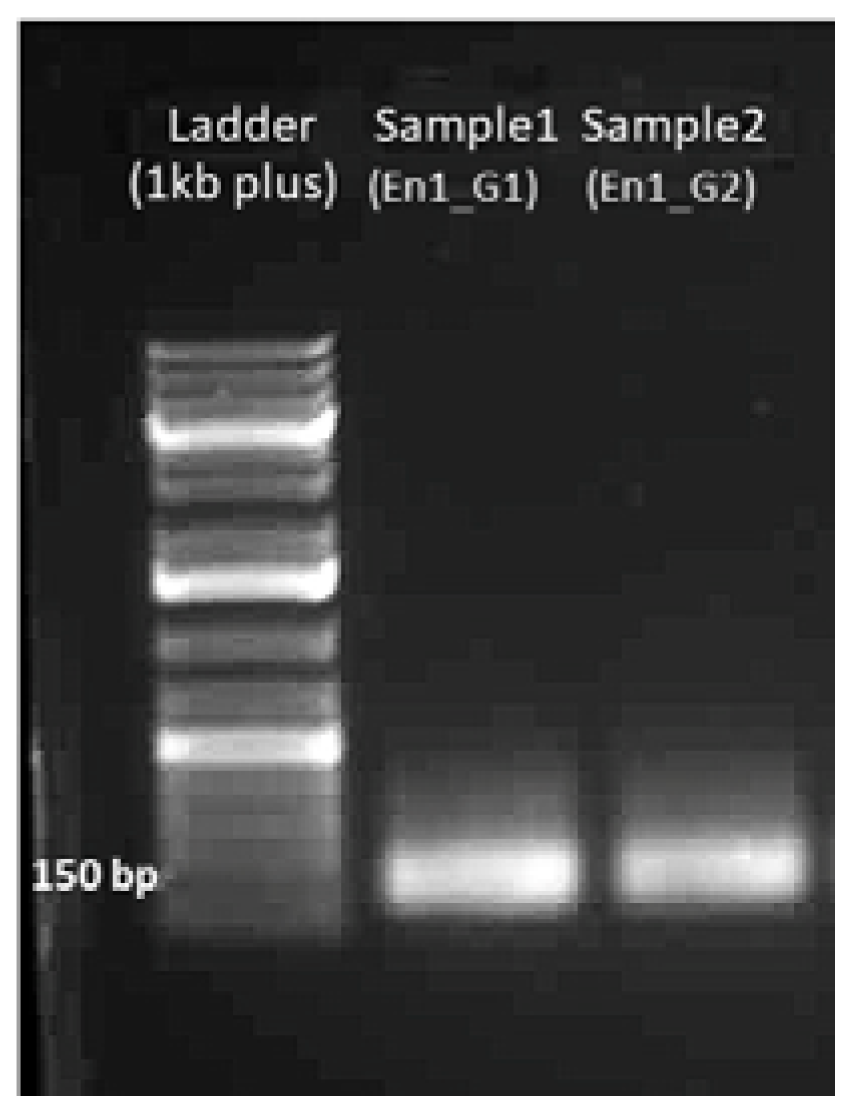
4.1.2. Gel Electrophoresis of sgDNA
4.1.3. Yield of sgRNA
4.1.4. Gel Electrophoresis of sgRNA
4.2. Preparation and Purification of Cas9 mRNA
4.3. Isolation of the Fragment of Interest (e.g., from Genomic DNA, Plasmid, etc.) and Verification of sgRNA via In Vitro Cleavage
4.3.1. Yield of Genomic DNA
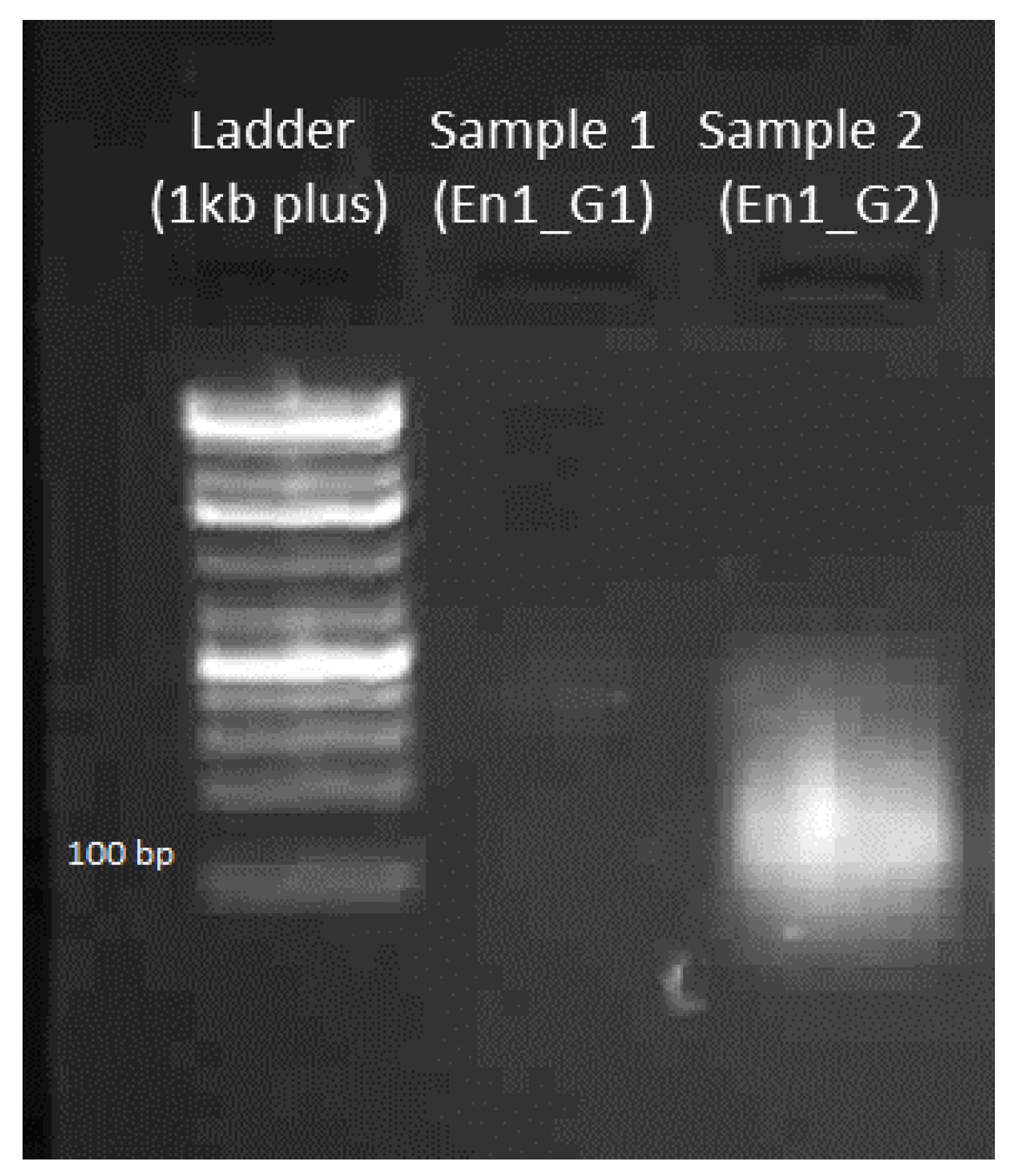
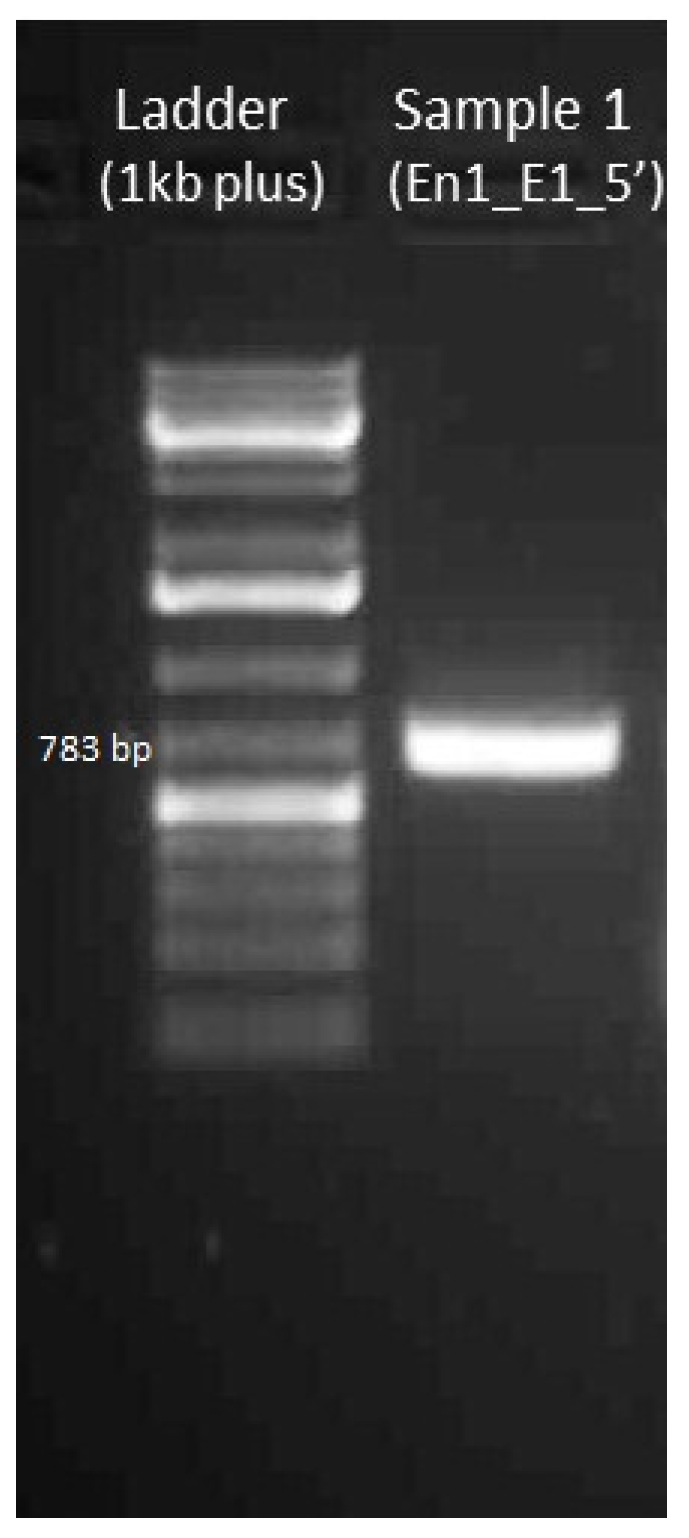
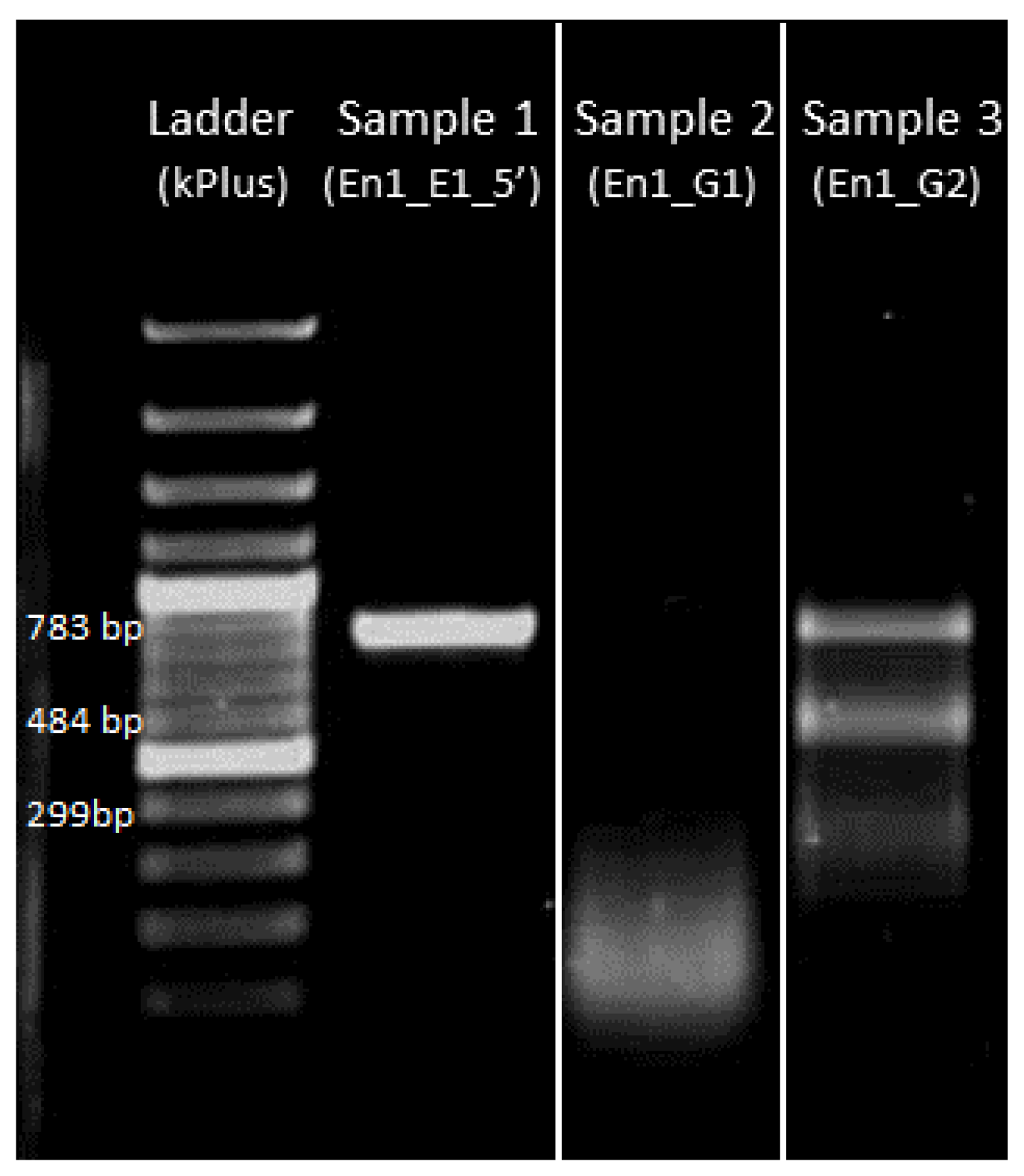
4.3.2. Gel Electrophoresis of Amplified DNA
4.3.3. Yield of Amplicon
4.3.4. Gel Electrophoresis of sgRNA-Cas9 Cleaved DNA Fragment
4.4. Embryo Collection, Needle Preparation, and Injection of the sgRNA-Cas9 Mixture
4.4.1. Number of Embryos
4.4.2. Thickness of Needle
4.4.3. Hatching Rate
4.4.4. Number of Mutants
4.5. T7 Endonuclease Assay and Genotyping of Mutants
4.5.1. Yield of DNA from Mutant Clones
4.5.2. Yield of DNA Amplicon
4.5.3. Gel Electrophoresis after T7 Endonuclease Assay
4.5.4. Number of Colonies
4.5.5. Yield of Plasmid
4.5.6. Sequencing Alignment Result
5. Reagents Setup
5.1. For sgRNA Design and In Vitro Verification
5.1.1. Preparation of 50× TAE buffer (1 L)
- In a 2 L beaker, add 500 mL MilliQ water and the reagents specified in Table 27.
- Transfer the content to a 1 L measuring cylinder. Raise the volume to one liter using MilliQ water.
- Mix the solution and transfer the content to a 1 L glass bottle.
- Autoclave the solution at 121 °C for 20 min and store the content at room temperature.Note: To prepare 1× TAE, add 20 mL of 50× TAE buffer and 980 mL of MilliQ water.
5.1.2. Preparation of 1% Agarose Gel
- Weigh 0.5 g of molecular grade agarose and transfer the content to a 200 mL glass bottle.
- Add 50 mL of 1× TAE buffer and heat the content inside microwave for 2 min.
- Remove the bottle and add 2 µL of SYBRSafe.
- Mix the content and pour on the gel tray.
5.1.3. Preparation of 3 M NaOAc (100 mL)
- Add 24.6 g of NaOAc in 60 mL MilliQ water.
- Adjust the pH using 1 N NaOH or 1N HCl.
- Transfer the content to a measuring cylinder and raise the volume to 100 mL with MilliQ.
- Store the content in a 200 mL glass bottle at room temperature.
5.1.4. Preparation of 70% Ethanol (100 mL)
- Add 30 mL of 100% ethanol into a 200 mL measuring cylinder and raise the volume to 100 mL using MilliQ water.
- Store the content in 200 mL glass bottle at room temperature.
5.2. For Cloning of Amplified DNA Fragments
5.2.1. Preparation of Ampicillin (100 mM)
- In a 20 mL measuring cylinder add 0.371 g of ampicillin and add molecular grade water up to a volume of 10 mL.
- Mix the content and make 1 mL aliquots in 1.5 mL microcentrifuge tubes. Store the content at −20 °C.
5.2.2. Preparation of IPTG (100 mM)
- In a 20 mL measuring cylinder, add 0.24 g of IPTG and raise the volume to 10 mL using molecular grade water.
- Mix the content and make 1 mL aliquots in 1.5 mL microcentrifuge tubes. Store the content at −20 °C.
5.2.3. Preparation of X-Gal (20 mg/mL)
- In a 20 mL measuring cylinder add 0.2 g of X-Gal and raise the volume to 10 mL using DMSO.
- Mix the content and make 1 mL aliquots in 1.5 mL microcentrifuge tubes. Store the content at −20 °C.
5.2.4. Preparation of LB Agar Plates and LB Broth
- For LB broth (100 mL)For LB agar (100 mL)
- Aliquot the LB broth in 50 mL glass tubes. Cotton plug the LB agar flask and LB broth tubes and autoclave at 121 °C for 20 min.
- LB broth tubes can be stored at room temperature or 4 °C for up to two months.
- Prepare LB agar plates by transferring 20 mL LB agar into Petri plates inside biological safety cabinet.
- LB agar plates can be stored at 4 °C for up to two months.
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Daimon, T.; Kiuchi, T.; Takasu, Y. Recent progress in genome engineering techniques in the silkworm, Bombyx mori. Dev. Growth Differ. 2014, 56, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; McLachlan, A.D.; Klug, A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. J. Trace Elem. Exp. Med. 2001, 14, 157–169. [Google Scholar] [CrossRef]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Beumer, K.J.; Trautman, J.K.; Bozas, A.; Liu, J.L.; Rutter, J.; Gall, J.G.; Carroll, D. Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc. Natl. Acad. Sci. USA 2008, 105, 19821–19826. [Google Scholar] [CrossRef] [PubMed]
- Takasu, Y.; Kobayashi, I.; Beumer, K.; Uchino, K.; Sezutsu, H.; Sajwan, S.; Carroll, D.; Tamura, T.; Zurovec, M. Targeted mutagenesis in the silkworm Bombyx mori using zinc finger nuclease mRNA injection. Insect Biochem. Mol. Biol. 2010, 40, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; McCammon, J.M.; Miller, J.C.; Faraji, F.; Ngo, C.; Katibah, G.E.; Amora, R.; Hocking, T.D.; Zhang, L.; Rebar, E.J.; et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.E.; Yeh, J.R.; Maeder, M.L.; Reyon, D.; Sander, J.D.; Peterson, R.T.; Joung, J.K. Rapid mutation of endogenous zebrafish genes using zinc finger nucleases made by Oligomerized Pool ENgineering (OPEN). PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Maeder, M.L.; Unger-Wallace, E.; Hoshaw, J.P.; Reyon, D.; Christian, M.; Li, X.; Pierick, C.J.; Dobbs, D.; Peterson, T.; et al. High frequency targeted mutagenesis in Arabidopsis thaliana using zinc finger nucleases. Proc. Natl. Acad. Sci. USA 2010, 107, 12028–12033. [Google Scholar] [CrossRef] [PubMed]
- Geurts, A.M.; Cost, G.J.; Freyvert, Y.; Zeitler, B.; Miller, J.C.; Choi, V.M.; Jenkins, S.S.; Wood, A.; Cui, X.; Meng, X.; et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 2009, 325, 433. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, J.; Petersen, B.; Santiago, Y.; Queisser, A.L.; Carnwath, J.W.; Lucas-Hahn, A.; Zhang, L.; Meng, X.; Gregory, P.D.; Schwinzer, R.; et al. Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc. Natl. Acad. Sci. USA 2011, 108, 15010. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Ochiai, H.; Sakuma, T.; Horch, H.W.; Hamaguchi, N.; Nakamura, T.; Bando, T.; Ohuchi, H.; Yamamoto, T.; Noji, S.; et al. Non-transgenic genome modifications in a hemimetabolous insect using zinc-finger and TAL effector nucleases. Nat. Commun. 2012, 3, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Merlin, C.; Beaver, L.E.; Taylor, O.R.; Wolfe, S.A.; Reppert, S.M. Efficient targeted mutagenesis in the monarch butterfly using zinc finger nucleases. Genome Res. 2012, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, L.; Martin, A.; Gibbs, M.; Braak, N.; Arif, S.; Breuker, C.J. CRISPR/Cas9 as the Key to Unlocking the Secrets of Butterfly Wing Pattern Development and Its Evolution [Internet], 1st ed.; Advances in Insect Physiology; Elsevier Ltd.: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 1509, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Smidler, A.L.; Terenzi, O.; Soichot, J.; Levashina, E.A.; Marois, E. Targeted Mutagenesis in the Malaria Mosquito Using TALE Nucleases. PLoS ONE 2013, 8, e74511. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, C.; Yu, Z.; Huang, P.; Wu, H.; Wei, C.; Zhu, N.; Shen, Y.; Chen, Y.; Zhang, B.; et al. Efficient and Specific Modifications of the Drosophila Genome by Means of an Easy TALEN Strategy. J. Genet. Genom. 2012, 39, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Takasu, Y.; Sajwan, S.; Daimon, T.; Osanai-Futahashi, M.; Uchino, K.; Sezutsu, H.; Tamura, T.; Zurovec, M. Efficient TALEN Construction for Bombyx mori Gene Targeting. PLoS ONE 2013, 8, e73458. [Google Scholar] [CrossRef]
- Takasu, Y.; Kobayashi, I.; Tamura, T.; Uchino, K.; Sezutsu, H.; Zurovec, M. Precise genome editing in the silkworm Bombyx mori using TALENs and ds- and ssDNA donors—A practical approach. Insect Biochem. Mol. Biol. 2016, 78, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.J.; Lo, T.W.; Zeitler, B.; Pickle, C.S.; Ralston, E.J.; Lee, A.H.; Amora, R.; Miller, J.C.; Leung, E.; Meng, X.; et al. Targeted genome editing across species using ZFNs and TALENs. Science 2011, 333, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, J.D.; Cade, L.; Khayter, C.; Reyon, D.; Peterson, R.T.; Joung, J.K.; Yeh, J.R. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol. 2011, 29, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.F.; Tan, W.; Lillico, S.G.; Stverakova, D.; Proudfoot, C.; Christian, M. Efficient TALEN-mediated gene knockout in livestock. Proc. Natl. Acad. Sci. USA 2012, 109, 17382–17387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Guo, X.; Liu, Y.; Cao, Y.; Deng, Y.; Chen, X.; Cheng, C.H.; Dawid, I.B.; Chen, Y.; Zhao, H. Efficient targeted gene disruption in Xenopus embryos using engineered transcription activator-like effector nucleases (TALENs). Proc. Natl. Acad. Sci. USA 2012, 109, 17484–17489. [Google Scholar] [CrossRef] [PubMed]
- Aryan, A.; Anderson, M.A.E.; Myles, K.M.; Adelman, Z.N. TALEN-Based Gene Disruption in the Dengue Vector Aedes aegypti. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Fujii, T.; Ishikawa, Y.; Matsuo, T. Targeted mutagenesis of an odorant receptor co-receptor using TALEN in Ostrinia furnacalis. Insect Biochem. Mol. Biol. 2016, 70, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Terns, M.P.; Terns, R.M. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 2011, 14, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, E.; Thakur, P.; Pareek, M.; Agarwal, N. Gene silencing by CRISPR interference in mycobacteria. Nat. Commun. 2015, 6, 6267. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Guo, Z.; Liu, Y.; Zhang, Y. Progress and prospects of CRISPR/Cas systems in insects and other arthropods. Front. Physiol. 2017, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R.; Tibbit, C.; Ponting, C.P.; Liu, J.L. Highly Efficient Targeted Mutagenesis of Drosophila with the CRISPR/Cas9 System. Cell Rep. 2013, 4, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hammond, A.; Galizi, R.; Kyrou, K.; Simoni, A.; Siniscalchi, C.; Katsanos, D.; Gribble, M.; Baker, D.; Marois, E.; Russell, S.; et al. A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat. Biotechnol. 2016, 34, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Koutroumpa, F.A.; Monsempes, C.; François, M.C.; De Cian, A.; Royer, C.; Concordet, J.P.; Jacquin-Joly, E. Heritable genome editing with CRISPR/Cas9 induces anosmia in a crop pest moth. Sci. Rep. 2016, 6, 29620. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Liu, Y.; Ai, D.; Jiang, X.; Dong, S.; Wang, G. A Pheromone Antagonist Regulates Optimal Mating Time in the Moth Helicoverpa armigera. Curr. Biol. 2017, 27, 1610–1615.e3. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, D.; Zhang, W.; Liu, G.; Zhang, L.; Zhao, L.; Fang, X.; Chen, L.; Dong, Y.; Chen, Y.; et al. Outbred genome sequencing and CRISPR/Cas9 gene editing in butterflies. Nat. Commun. 2015, 6, 8212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Reed, R.D. Genome editing in butterflies reveals that spalt promotes and Distal-less represses eyespot colour patterns. Nat. Commun. 2016, 7, 11769. [Google Scholar] [CrossRef] [PubMed]
- Connahs, H.; Tlili, S.; van Creij, J.; Loo, T.Y.; Banerjee, T.; Saunders, T.E.; Monteiro, A. Disrupting different Distal-less exons leads to ectopic and missing eyespots accurately modeled by reaction-diffusion mechanisms. bioRxiv 2017, 2, 183491. [Google Scholar] [CrossRef]
- Prakash, A.; Monteiro, A. apterous A specifies dorsal wing patterns and sexual traits in butterflies. Proc. R. Soc. B Biol. Sci. 2018, 285. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Monteiro, A. Melanin pathway genes regulate color and morphology of butterfly wing scales. Mech. Dev. 2017, 145, S109. [Google Scholar] [CrossRef]
- Zhang, L.; Martin, A.; Perry, M.W.; van der Burg, K.R.; Matsuoka, Y.; Monteiro, A.; Reed, R.D. Genetic Basis of Melanin Pigmentation in Butterfly Wings. Genetics 2017, 205, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liang, D.; Wang, Y.; Bai, M.; Tang, W.; Bao, S.; Yan, Z.; Li, D.; Li, J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 2013, 13, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dai, Z.; Liang, Y.; Yin, M.; Ma, K.; He, M.; Ouyang, H.; Teng, C.B. Sequence-specific inhibition of microRNA via CRISPR/CRISPRi system. Sci. Rep. 2014, 4, 3943. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Beldade, P.; Peralta, C.M. Developmental and evolutionary mechanisms shaping butterfly eyespots. Curr. Opin. Insect. Sci. 2017, 19, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A. Origin, Development, and Evolution of Butterfly Eyespots. Annu. Rev. Entomol. 2015, 60, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Reed, R.D. A Practical Guide to CRISPR/Cas9 Genome Editing in Lepidoptera. In Divers. Evol. Butterfly Wing Patterns; Sekimura, T., Nijhout, H., Eds.; Springer: Singapore, 2017; Volume 99, pp. 155–172. [Google Scholar]
- Özsu, N.; Monteiro, A. Wound healing, calcium signaling, and other novel pathways are associated with the formation of butterfly eyespots. BMC Genom. 2017, 18, 788. [Google Scholar] [CrossRef] [PubMed]
- Saenko, S.V.; French, V.; Brakefield, P.M.; Beldade, P. Conserved developmental processes and the formation of evolutionary novelties: Examples from butterfly wings. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 1549–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keys, D.N. Recruitment of a hedgehog Regulatory Circuit in Butterfly Eyespot Evolution. Science 1999, 283, 532–534. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, C.R.; Selegue, J.E.; Monteiro, A.; French, V.; Brakefield, P.M.; Carroll, S.B. The generation and diversification of butterfly eyespot color patterns. Curr. Biol. 2001, 11, 1578–1585. [Google Scholar] [CrossRef]
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2015, 31, 1120–1123. [Google Scholar] [CrossRef] [PubMed]
- Challis, R.J.; Kumar, S.; Dasmahapatra, K.K.; Jiggins, C.D.; Blaxter, M. Lepbase: The Lepidopteran genome database. bioRxiv. 2016, 56994. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 2007, 35, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Sutter Instrument. Pipette Cookbook. 2018. Available online: https://www.sutter.com/PDFs/pipette_cookbook.pdf (accessed on 4 May 2018).
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Stages | Time for Completion |
|---|---|
| 1. Design, synthesis and verification of sgRNA | 3 days |
| 1.1. Design and ordering oligonucleotides | 1 day |
| 1.2. Synthesis and purification of sgDNA | 3 h |
| 1.3. Synthesis of sgRNA | 18 h |
| 1.4. Purification of sgRNA | 1 h |
| 1.5. In vitro verification of sgRNA | 4 h |
| 2. Injection of sgRNA-Cas9 mixture into embryos | 1 day |
| 2.1. Preparation of sgRNA-Cas9 mixture | 15 min |
| 2.2. Needle preparation | 5 min |
| 2.3. Collection of embryos | 1 h |
| 2.4. Arranging embryos in double-sided tape | 15 min |
| 2.5. Injection of sgRNA-Cas9 mixture | 1 h |
| 2.6. Incubation of injected embryos | 10 min |
| 3. Rearing of hatched individuals | 4–5 weeks |
| 3.1. Embryonic stage | 3–4 days |
| 3.2. Larval stage | 3 weeks |
| 3.3. Pupal stage | 1–2 weeks |
| 4. Genotyping of mutant individuals | 3 days |
| 4.1. Isolating DNA from mutant clones | 1 day |
| 4.2. Amplification of region of interest | 3 h |
| 4.3. T7 endonuclease assay on the region of interest | 2 h |
| 4.4. Cloning of the region of interest | 1 day |
| 4.5. Isolating plasmids from clones | 1 h |
| 4.6. Sequencing region of interest | 5 h |
| 4.7. Analyzing the sequencing data | 1–2 h |
| ● Synthesis of Cas9 mRNA (optional) | 1 day |
| Primer | Sequence |
|---|---|
| Forward primer for En1_guide1(En1_G1) | GAAATTAATACGACTCACTATAGGAGACCGTTGCAGTCCGAAC CGTTTTAGAGCTAGAAATAGC |
| Forward primer for En1_guide2(En2_G2) | GAAATTAATACGACTCACTATAGGGTTTGACAGAACGATGACA GGTTTTAGAGCTAGAAATAGC |
| Reverse primer for guide synthesis (CRISPR_R) | AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACT AGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC |
| Primer | Sequence |
|---|---|
| En1_Exon1_with_5′UTR_Forward (En1_E1_5′_F) | GCGCATCGTGTTGTCAAAAA |
| En1_Exon1_with_5′UTR_Reverse (En1_E1_5′_R) | TAGATTGCTGTTCCCGCTTT |
| M13F | CGCCAGGGTTTTCCCAGTCACGAC |
| M13R | TCACACAGGAAACAGCTATGAC |
| Reagents | Volume (µL) |
|---|---|
| Q5 high fidelity DNA polymerase | 1 |
| Q5 polymerase buffer 10× | 20 |
| dNTP mix | 2 |
| Forward Primer (En1_G1) | 3 |
| Reverse Primer (CRISPR_R) | 3 |
| Molecular grade water | 71 |
| Temperature (°C) | Time (s) | Number of Cycles |
|---|---|---|
| 98 | 30 | 1 |
| 98 | 30 | |
| 57 | ||
| 72 | ||
| 72 | 120 | 1 |
| 4 | ∞ | 1 |
| Reagents | Volume (µL) |
|---|---|
| T7 10× buffer | 2 |
| T7 RNA polymerase | 2 |
| ATP, GTP, UTP, CTP (10 mM) | 8 (2 µL each) |
| sgDNA (500 ng/µL) | 2 |
| Ribolock | 0.5 |
| Molecular grade water | 5.5 |
| Reagents | Volume (µL) |
|---|---|
| Template DNA | 2 |
| 2× NTP (provided in the mMESSAGE mMACHINE kit) | 10 |
| 10× buffer (provided in the mMESSAGE mMACHINE kit) | 2 |
| T3 enzyme (provided in the mMESSAGE mMACHINE kit) | 2 |
| Ribolock | 0.5 |
| Molecular grade water | 3.5 |
| Reagents | Volume (µL) |
|---|---|
| 10 mM ATP (provided in Poly(A) Tailing kit) | 10 |
| 25 mM MnCl2 (provided in Poly(A) Tailing kit) | 10 |
| 2× buffer (provided in Poly(A) Tailing kit) | 20 |
| E-PAP (provided in Poly(A) Tailing kit) | 4 |
| Molecular grade water | 30 |
| Reagents | Volume (µL) |
|---|---|
| 2× PCRBIO Taq Mix Red | 12.5 |
| Forward Primer (En1_E1_5′_F) | 1 |
| Reverse Primer (En1_E1_5′_R) | 1 |
| Template (gDNA/plasmid/cDNA) | 1 |
| Molecular grade water | 9.5 |
| Temperature (°C) | Time (s) | Number of Cycles |
|---|---|---|
| 95 | 60 | 1 |
| 95 | 40 | |
| Gradient (55–65) | ||
| 72 | ||
| 4 | ∞ | 1 |
| Reagents | Volume (µL) |
|---|---|
| Cas9 buffer 10× | 1 |
| Cas9 Protein NLS (3300 ng/µL) | 0.1 |
| sgRNA (600 ng/µL) | 0.5 |
| Molecular grade water | 8.4 |
| Reagents | Volume (µL) |
|---|---|
| NEB buffer 3.1 | 1 |
| sgRNA (600 ng/µL) | 0.5 |
| Molecular grade water | 8.4 |
| a | |
| Conditions | Value |
| Heat | 625 |
| Pull | 10 |
| Vel | 10 |
| Time | 250 |
| b | |
| Conditions | Value |
| Heat | 625 |
| Pull | 10 |
| Vel | 10 |
| Time | 200 |
| c | |
| Conditions | Value |
| Heat | 625 |
| Pull | 10 |
| Vel | 10 |
| Time | 150 |
| Reagents | Volume (µL) |
|---|---|
| Cas9 buffer 10× | 1 |
| Cas9 Protein NLS (3300 ng/µL) | 1 |
| sgRNA (600 ng/µL) | 5 |
| Molecular grade water | 3 |
| Reagents | Volume (µL) |
|---|---|
| 2× PCRBIO Taq Mix Red | 12.5 |
| Forward Primer (En1_E1_5′_F) | 1 |
| Reverse Primer (En1_E1_5′_R) | 1 |
| Template (gDNA/plasmid/cDNA) | 1 |
| Molecular grade water | 9.5 |
| Temperature (°C) | Time (s) | Number of Cycles |
|---|---|---|
| 95 | 60 | 1 |
| 95 | 40 | |
| Gradient (55–65) | ||
| 72 | ||
| 4 | ∞ | 1 |
| Reagents | Volume (µL) |
|---|---|
| Amplified DNA (200 ng/µL) | 1 |
| 10× NEB Buffer 2 | 2 |
| Molecular grade water | 17 |
| Temperature (°C) | Time (s) |
|---|---|
| 95 | 300 |
| 95–85 | 5 |
| 85–25 | 600 |
| 8 | ∞ |
| Reagents | Volume (µL) |
|---|---|
| 2× Rapid ligation buffer | 5 |
| pGEM-T vector | 0.5 |
| Amplified DNA (200 ng/µL) | 0.5 |
| T4 DNA ligase | 1 |
| Molecular grade water | 3 |
| Reagents | Volume (µL) |
|---|---|
| IPTG | 25 |
| X-GAL | 25 |
| Ampicillin | 25 |
| Reagents | Volume (µL) |
|---|---|
| 2× PCRBIO Taq Mix Red | 12.5 |
| M13F primer | 1 |
| M13R primer | 1 |
| Homogenized clone | 1 |
| Molecular grade water | 9.5 |
| Temperature (°C) | Time (s) | Number of Cycles |
|---|---|---|
| 95 | 60 | 1 |
| 95 | 30 | |
| 57 | ||
| 72 | ||
| 4 | ∞ | 1 |
| Reagents | Volume (µL) |
|---|---|
| Plasmid | 1 |
| M13F primer | 3 |
| BigDye Terminator v3.1 | 0.5 |
| 5× BigDye buffer | 2 |
| Molecular Grade Buffer | 3.5 |
| Reagents | Volume (µL) |
|---|---|
| Plasmid | 1 |
| M13R primer | 3 |
| BigDye Terminator v3.1 | 0.5 |
| 5× BigDye buffer | 2 |
| Molecular Grade Buffer | 3.5 |
| Temperature (°C) | Time (s) | Number of Cycles |
|---|---|---|
| 96 | 60 | 1 |
| 96 | 30 | |
| 54 | ||
| 60 | ||
| 4 | ∞ | 1 |
| Experiment | Gene (Date/Month) | Eggs Injected | Hatchlings | % Hatching |
|---|---|---|---|---|
| 1 | En1 (4 August 2017) 300 ng/µL with Cas9 protein | 280 | 16 | 5.71 |
| 2 | En1 (22 October 2017) 100 ng/µL with Cas9 protein | 187 | 18 | 9.63 |
| 3 | En1 (23 September 2017) 300 ng/µL with Cas9 protein | 272 | 35 | 12.86 |
| 4 | En1 (15 February 2018) 300 ng/µL with Cas9 mRNA | 164 | 14 | 8.54 |
| 5 | En1 (1 March 2018) 300 ng/µL with Cas9 protein | 352 | 168 | 47.72 |
| Reagents | Weight/Volume |
|---|---|
| Trizma base | 242 g |
| Disodium EDTA | 18.61 g |
| Glacial Acetic Acid | 57.1 mL |
| Reagents | Weight/Volume |
|---|---|
| LB base | 2.5 g |
| MilliQ water | 100 mL |
| Reagents | Weight/Volume |
|---|---|
| LB agar | 3.2 g |
| MilliQ water | 100 ml |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, T.D.; Monteiro, A. CRISPR-Cas9 Mediated Genome Editing in Bicyclus anynana Butterflies. Methods Protoc. 2018, 1, 16. https://doi.org/10.3390/mps1020016
Banerjee TD, Monteiro A. CRISPR-Cas9 Mediated Genome Editing in Bicyclus anynana Butterflies. Methods and Protocols. 2018; 1(2):16. https://doi.org/10.3390/mps1020016
Chicago/Turabian StyleBanerjee, Tirtha Das, and Antónia Monteiro. 2018. "CRISPR-Cas9 Mediated Genome Editing in Bicyclus anynana Butterflies" Methods and Protocols 1, no. 2: 16. https://doi.org/10.3390/mps1020016
APA StyleBanerjee, T. D., & Monteiro, A. (2018). CRISPR-Cas9 Mediated Genome Editing in Bicyclus anynana Butterflies. Methods and Protocols, 1(2), 16. https://doi.org/10.3390/mps1020016