
A Robust Expression and Purification Method for Production of SpCas9-GFP-MBP Fusion Protein for In Vitro Applications



Abstract
:1. Introduction

| Plasmid | Reported Yield/L of Bacterial Culture | Reference |
|---|---|---|
| pMJ915 (MBP−Cas9) | 1.77 mg | [15] |
| pET-NLS Cas9−6xHis | 1.2 mg | [22] |
| pET-28b-Cas9−His (Cas9−His) | 8.38 mg (best condition reported) | [23] |
| pET15 (Cas9−6xHis) | 6 mg | [24] |
| His−MBP−Cas9 Cas9 D10A, H840A and D10A/H840A point mutants | NR * | [16,17] |
| PMJ922 (His−MBP−TEV−Cas9 NLS−GFP) | NR * | [18] |
| His−MBP−Cas9 Cas9 D10A, H840A and D10A/H840A point mutants | NR * | [19,20] |
| D10A/H840A dCas9 | NR * | [21] |
| pET-Cas9 NLS−6xHis | NR * | [29] |
| pET-28b-Cas9−His | NR * | [30] |
| pET-28b-Cas9−His | NR * | [31] |
2. Experimental Design
2.1. Materials
- pMJ922 (Addgene, Cambridge, MA, USA; plasmid no. 78312).
- Escherichia coli BL21 (DE3) pLysS (ThermoFisher Scientific, Waltham, MA, USA; Cat. no.: C602003).
- TRIS (Duchefa Biochemie; Haarlem, The Netherlands; Cat. no.: T1501).
- HEPES (Sigma-Aldrich, St. Louis, MO, USA; Cat. no.: H-3375).
- Potassium chloride (Sigma-Aldrich; St. Louis, MO, USA; Cat. no.: P3911).
- Magnesium chloride hexahydrate (Sigma-Aldrich; St. Louis, MO, USA; Cat. no.: M2670).
- Sodium chloride (Sigma-Aldrich St. Louis, MO, USA; Cat. no.: S7653).
- Tris(2-carboxyethyl)phosphine hydrochloride (TCEP; Sigma-Aldrich; St. Louis, MO, USA; Cat. no.: C4706).
- DTT (Duchefa Biochemie; Haarlem, The Netherlands;Cat. no.: D1309).
- Imidazole (Sigma-Aldrich; St. Louis, MO, USA; Cat. no.: I-2399).
- Glycerol (Sigma-Aldrich; St. Louis, MO, USA; Cat. no.: G5516).
- Benzonase (Pierce, Waltham, MA, USA; Cat. no. 88701).
- Protease inhibitor tablets (Roche, Basel, Switzerland; Cat. no. 05892970001).
- HiTrap IMAC FF (GE Healthcare; Chicago, IL, USA; Cat. no. 17092104).
- PD-10 desalting columns (GE Healthcare; Cat. no. 17-0851-01).
- HiTrap SP HP (GE Healthcare; Chicago, IL, USA; Cat. no. GE29-0513-24).
- Polycarbonate bottles (Beckman, Brea, CA, USA; Cat. no. 355605).
2.2. Equipment
- Refrigerated orbital shaker (Labotech, Midrand, South Africa; Model: ZWY-211C).
- Magnetic stirrer (Thermolyne, Waltham, MA, USA).
- Sonicator (Benchmark Scientific, Sayreville, NJ, USA. Model: Pulse 150).
- Beckman J2HC refrigerated centrifuge (Beckman, Brea, CA, USA; Cat. no.: 8043-30-1105).
- Beckman JA-10 rotor (Beckman, Brea, CA, USA; Cat. no.: 369687).
- Beckman JA-20 rotor (Beckman, Brea, CA, USA; Cat. no.: 334831).
- ÄKTA FPLC system (GE Healthcare, Uppsala, Sweden; Cat. no.: 29-0598-78 AB).
3. Procedure
3.1. Protein Expression
- Transform pMJ922 into competent E. coli BL21 (DE3) pLysS. Isolate single colonies and cultivate them for 8 h in liquid LB containing 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. Prepare stocks using 25% glycerol final concentration.
![Mps 05 00044 i001]() CRITICAL STEP: overgrowing bacterial cells may lead to plasmid loss. It is recommendable to check plasmid presence at this step either through PCR or miniprep.
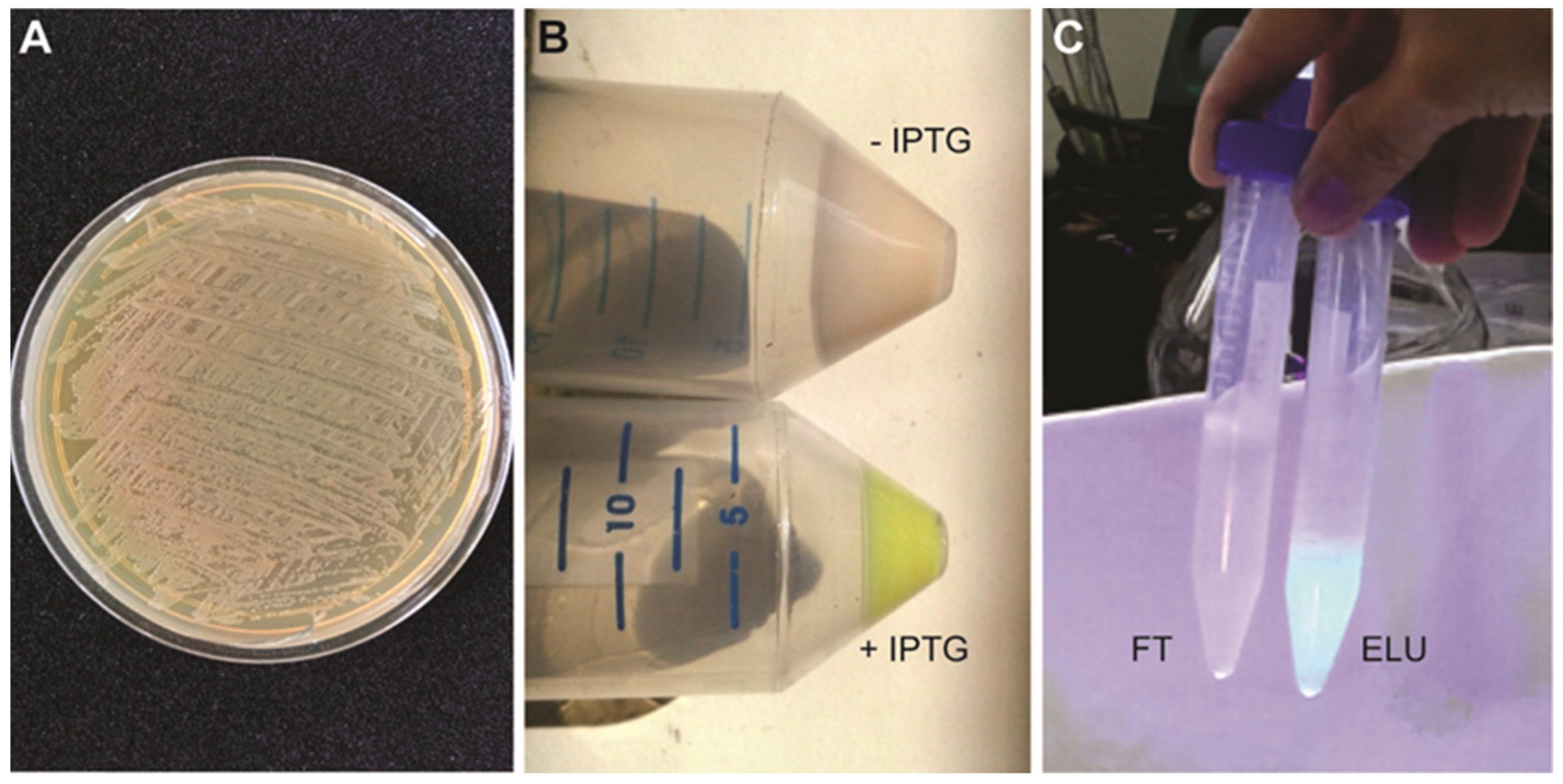
CRITICAL STEP: overgrowing bacterial cells may lead to plasmid loss. It is recommendable to check plasmid presence at this step either through PCR or miniprep. - Prepare starting plates for protein expression. Spread glycerol stock onto a solid LB plate containing 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. Completely streak the plate to cover the entire plate with bacteria. Incubate at 37 °C overnight (Figure 2A).
![Mps 05 00044 i001]() CRITICAL STEP: this plate is used in replacement of an overnight grown culture. Prepare two plates for this purpose.
CRITICAL STEP: this plate is used in replacement of an overnight grown culture. Prepare two plates for this purpose. - Using a sterile spatula, scrape all the bacteria from a plate and resuspend the cells in 5 mL LB media. Add this suspension to a 2 L Erlenmeyer containing 500 mL TB media (24 g/L yeast extract, 20 g/L tryptone, 4 mL/L glycerol, 17 mM KH2PO4, 72 mM K2HPO4) supplemented with 200 μg/mL ampicillin and 34 μg/mL chloramphenicol. Incubate at 37 °C and 200 rpm until absorbance at 600 nm reach 0.6. Prepare two Erlenmeyers for this purpose.
- Cool down the cultures at 4 °C for at least 30 min to arrest growth.
- Supplement the cultures with ampicillin once again (to reach a final concentration of 400 μg/mL) and subsequently induce protein expression by adding 200 μM isopropyl-D-1-thiogalactopyranoside (IPTG). Cultivate for 16 h at 18 °C and 180 rpm.
![Mps 05 00044 i001]() CRITICAL STEP: ampicillin reinforcement ensures that selection of the plasmid is maintained during overnight induction.
CRITICAL STEP: ampicillin reinforcement ensures that selection of the plasmid is maintained during overnight induction. - Harvest the cells in four 500 mL polycarbonate bottles (Beckman, Cat. no. 355605) by centrifugation at 3000× g for 20 min at 4 °C. Resuspend the cell pellet in 100 mL of the supernatant and transfer to two 50 mL Falcon tubes. Centrifuge at 3000× g for 20 min at 4 °C and weight the pellet.
![Mps 05 00044 i001]() CRITICAL STEP: the pellet should be greenish due to the GFP expression (Figure 2B).
CRITICAL STEP: the pellet should be greenish due to the GFP expression (Figure 2B). ![Mps 05 00044 i002]() PAUSE STEP: pellets can be stored at −80 °C for several months until use.
PAUSE STEP: pellets can be stored at −80 °C for several months until use.


{kind=link}
{kind=link}
{kind=link}
{kind=link}

3.2. Protein Purification
3.2.1. Bacterial Lysis
- Resuspend 3–6 g of cell pellet in IMAC buffer A (50 mM TRIS pH 7.5, 500 mM NaCl, 5 mM MgCl2) in a 5:1 buffer:pellet ratio. Perform an initial lysis step by three cycles of freezing/thawing the pellet at −80 °C. Transfer the suspension to a small beaker and add lysozyme to a final concentration of 0.1 mg/mL, 1 mM DTT final concentration, additional MgCl2, 1:500 v/v from 1 M stock solution, a protease inhibitor tablet (Roche, Cat. no. 05892970001) and 1 μL of benzonase (25 kU/μL, Pierce, Cat. 88701).
![Mps 05 00044 i001]() CRITICAL STEP: using benzonase instead of DNase greatly simplifies further purification steps. Benzonase digests all forms of nucleic acids, which can affect purity during IMAC and binding to the ion exchange resin if left undigested.Contamination with nucleic acids from E. coli can be problematic for Cas9 purification because these molecules can interfere with the binding capacity of Cas9 to the IEX resin due to PI alteration. This problem can be addressed by using RNase in addition to DNase or, alternatively, benzonase, which exhibits both DNase and RNase activity.
CRITICAL STEP: using benzonase instead of DNase greatly simplifies further purification steps. Benzonase digests all forms of nucleic acids, which can affect purity during IMAC and binding to the ion exchange resin if left undigested.Contamination with nucleic acids from E. coli can be problematic for Cas9 purification because these molecules can interfere with the binding capacity of Cas9 to the IEX resin due to PI alteration. This problem can be addressed by using RNase in addition to DNase or, alternatively, benzonase, which exhibits both DNase and RNase activity. - Incubate for 30 min on ice under very gentle stirring to avoid foam formation.
- Perform 4 cycles of sonication using 40% amplitude pulses for 3 min, with pauses of 2 min in between them.
- Clarify the suspension by centrifugation for 15 min at 16,000× g at 4 °C. Transfer the supernatant to a new tube and centrifuge again for 45 min at 16,600× g and 4 °C.
- OPTIONAL STEP: filtrate through a 0.45 μm filter if the lysate is not totally transparent.
3.2.2. IMAC
- 6.
- Load the soluble fraction into a 5 mL IMAC HiTrap FastFlow column (GE, Cat. 17092104), previously equilibrated with 10 column volumes (CV) of IMAC buffer A. Wash the column with at least 25 CV of IMAC buffer A. Elute resin-bound proteins with IMAC buffer B (50 mM TRIS pH 7.5, 250 mM NaCl, 5 mM MgCl2, 400 mM imidazole) in 0.5 mL fractions.
![Mps 05 00044 i001]() CRITICAL STEP: extensively wash unbound protein. More than 25 CV may be needed in some cases. Measure the absorbance of IMAC buffer A and wash until this value is reached again.
CRITICAL STEP: extensively wash unbound protein. More than 25 CV may be needed in some cases. Measure the absorbance of IMAC buffer A and wash until this value is reached again.
3.2.3. Buffer Exchange in PD-10
- 7.
- Pool the protein containing fractions and perform buffer exchange in PD-10 column (GE, Cat. 17-0851-01) following the manufacturer’s instructions. Use ion exchange (IEX) buffer A (50 mM TRIS pH 7.5, 100 mM NaCl, 5 mM MgCl2) for equilibration. After elution, add DTT to 1 mM final concentration. For in vitro applications, continue in Section 3.2.4.
- 8.
- OPTIONAL STEP: For in vivo applications, it is recommended to remove the 6xHis−MBP tag by cleavage with TEV-P (see Section 3.2.7).
3.2.4. IEX
- 9.
- Load the protein into the cation exchange SpHp HighTrap column (GE, Cat. GE29-0513-24) previously equilibrated with IEX buffer A. Wash the column with at least 25 CV of IEX buffer A. Elute the protein with a linear gradient of 0.1–1 M NaCl in 20 CV, collecting in fractions of 0.5 mL.OPTIONAL STEP: This step can be performed using a peristaltic pump, in which case elution is performed in a single step with IEX buffer B (50 mM TRIS pH 7.5, 1 M NaCl, 5 mM MgCl2). The purity obtained is not as good compared to the gradient elution, but the protein can still be used for in vitro cleavage assays.
3.2.5. Buffer Exchange in PD-10
- 10.
- Pool the protein containing fractions and perform buffer exchange in a PD-10 column equilibrated in storage buffer (30 mM HEPES pH 7.5, 200 mM NaCl, 2 mM MgCl2, and 1 mM TCEP).
- 11.
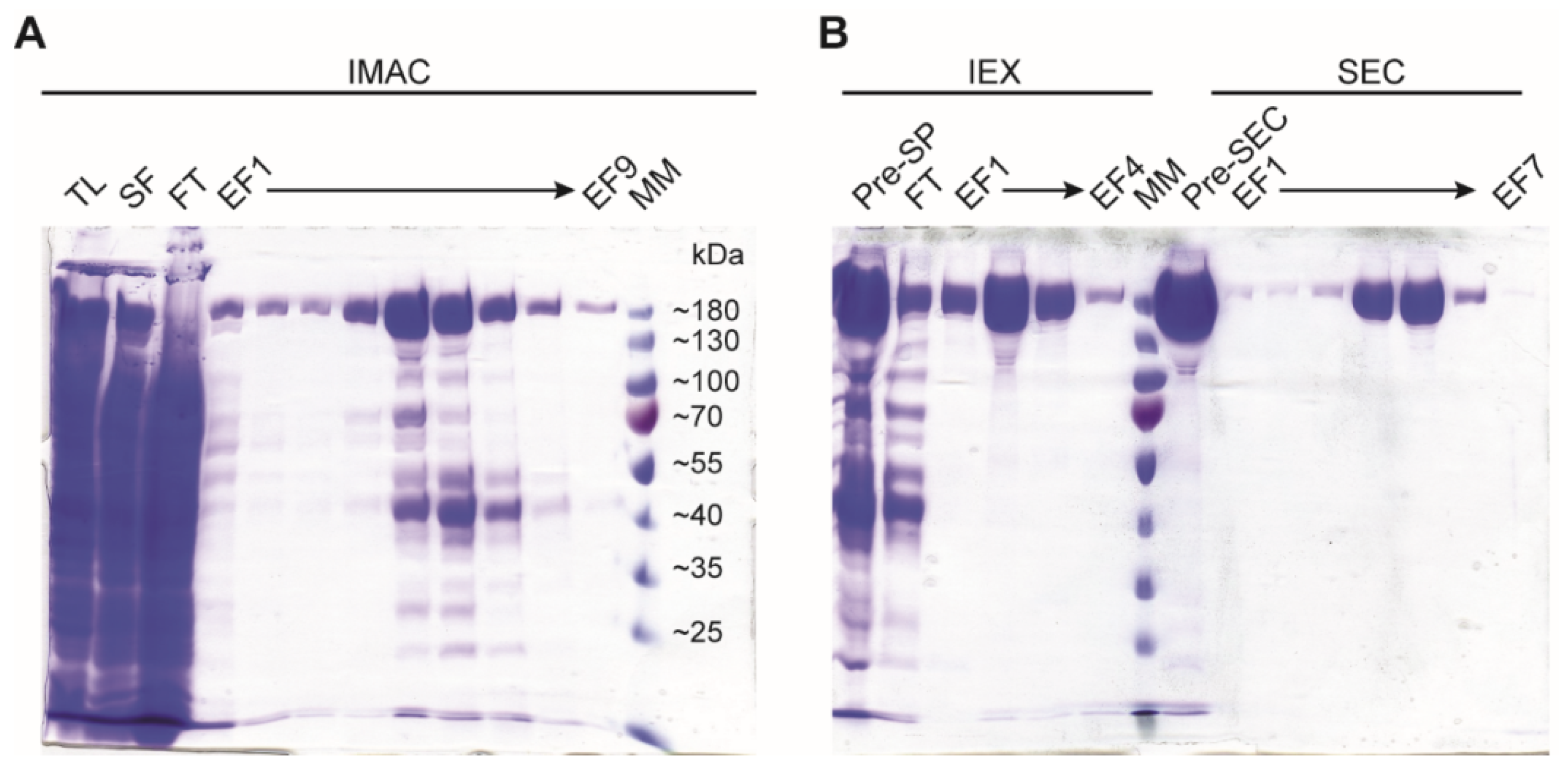
- OPTIONAL STEP: If greater purity is needed, use a Superdex 200 16/600 size exclusion column (GE Healthcare), equilibrated in storage buffer for the final polishing step (Figure 3).
3.2.6. SDS-PAGE
- 12.
- Check the purity of the purified protein by SDS-PAGE in 10% acrylamide-bisacrylamide (29:1) gel (Figure 3).
3.2.7. Removal of His−MBP Tag for In Vivo Applications (Optional Step 15, Expanded)
3.2.8. Protein Storage
- 13.
- Quantify the protein concentration using theoretical ξ280 = 211,920 M−1 cm−1.
- 14.
- Store the protein at a final concentration > 1 mg/mL (usually the protein concentration is higher than 1 mg/mL after buffer exchange). Prepare single use aliquots and flash freeze in liquid nitrogen. Store at −80 °C. The protein remains active for over a year.
3.3. In Vitro Nuclease Activity Assay
- Prepare 50 μL of reaction mixture in 1X Cas9 buffer (0.2 mM HEPES pH 7.5, 1.5 mM KCl, 0.1 mM MgCl2, 10 μM EDTA, 50 μM DTT freshly added) as indicated in Table 3.
- Allow the reaction to equilibrate for 15 min at room temperature. Add 300 ng of the PCR product, briefly vortex and spin. Incubate at 37 °C for 2 h.
- Add 1 μL of 20 mg/mL proteinase K and incubate for 10 min at 37 °C. Analyze in agarose gel. The agarose percentage should be according to the expected DNA fragment sizes after cleavage. An example of a typical activity assay is shown in Figure 4.
4. Expected Results
5. Protocol Alternatives and Troubleshooting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wright, A.V.; Nuñez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Brant, E.; Budak, H.; Zhang, B. CRISPR/Cas: A Nobel Prize award-winning precise genome editing technology for gene therapy and crop improvement. J. Zhejiang Univ. Sci. B 2021, 22, 253–284. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gao, L.; Feng, W.; Guo, C.; Yang, Q.; Li, F.; Le, X.C. The CRISPR–Cas toolbox for analytical and diagnostic assay development. Chem. Soc. Rev. 2021, 50, 11844–11869. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef]
- Huang, T.P.; Newby, G.A.; Liu, D.R. Precision genome editing using cytosine and adenine base editors in mammalian cells. Nat. Protoc. 2021, 16, 1089–1128. [Google Scholar] [CrossRef]
- Atkins, P.A.; Voytas, D.F. Overcoming bottlenecks in plant gene editing. Curr. Opin. Plant Biol. 2020, 54, 79–84. [Google Scholar] [CrossRef]
- Banakar, R.; Schubert, M.; Kurgan, G.; Rai, K.M.; Beaudoin, S.F.; Collingwood, M.A.; Vakulskas, C.A.; Wang, K.; Zhang, F. Efficiency, Specificity and Temperature Sensitivity of Cas9 and Cas12a RNPs for DNA-free Genome Editing in Plants. Front. Genome Ed. 2022, 3, 760820. [Google Scholar] [CrossRef]
- Woo, J.W.; Kim, J.; Kwon, S.I.; Corvalán, C.; Cho, S.W.; Kim, H.; Kim, S.-G.; Kim, S.-T.; Choe, S.; Kim, J.-S. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat. Biotechnol. 2015, 33, 1162–1164. [Google Scholar] [CrossRef]
- Malnoy, M.; Viola, R.; Jung, M.-H.; Koo, O.-J.; Kim, S.; Kim, J.-S.; Velasco, R.; Kanchiswamy, C.N. DNA-Free Genetically Edited Grapevine and Apple Protoplast Using CRISPR/Cas9 Ribonucleoproteins. Front. Plant Sci. 2016, 7, 1904. [Google Scholar] [CrossRef]
- Subburaj, S.; Chung, S.J.; Lee, C.; Ryu, S.-M.; Kim, D.H.; Kim, J.-S.; Bae, S.; Lee, G.-J. Site-directed mutagenesis in Petunia × hybrida protoplast system using direct delivery of purified recombinant Cas9 ribonucleoproteins. Plant Cell Rep. 2016, 35, 1535–1544. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, K.; Li, T.; Zhang, Y.; Wang, Y.; Zhao, Q.; Liu, J.; Zhang, H.; Liu, C.; Ran, Y.; et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nat. Commun. 2017, 8, 14261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, Z.; Zong, Y.; Wang, Y.; Liu, J.; Chen, K.; Qiu, J.-L.; Gao, C. Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat. Commun. 2016, 7, 12617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, M.; Turesson, H.; Olsson, N.; Fält, A.-S.; Ohlsson, P.; Gonzalez, M.N.; Samuelsson, M.; Hofvander, P. Genome editing in potato via CRISPR-Cas9 ribonucleoprotein delivery. Physiol. Plant. 2018, 164, 378–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metje-Sprink, J.; Menz, J.; Modrzejewski, D.; Sprink, T. DNA-Free Genome Editing: Past, Present and Future. Front. Plant Sci. 2019, 9, 1957. [Google Scholar] [CrossRef]
- Dilworth, D.; Harding, R.; Ravichandran, M.; Barsyte-Lovejoy, D. Purification of Recombinant Cas9. Available online: https://openlabnotebooks.org/purification-of-recombinant-cas9/ (accessed on 2 February 2022).
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [Green Version]
- Burger, A.; Lindsay, H.; Felker, A.; Hess, C.; Anders, C.; Chiavacci, E.; Zaugg, J.; Weber, L.M.; Catena, R.; Jinek, M.; et al. Maximizing mutagenesis with solubilized CRISPR-Cas9 ribonucleoprotein complexes. Development 2016, 143, 2025–2037. [Google Scholar] [CrossRef] [Green Version]
- Anders, C.; Jinek, M. Vitro Enzymology of Cas9. Methods Enzymol. 2014, 546, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, N.; Kagale, S.; Bhowmik, P.; Song, H. A Two-Step Method for Obtaining Highly Pure Cas9 Nuclease for Genome Editing, Biophysical, and Structural Studies. Methods Protoc. 2018, 1, 17. [Google Scholar] [CrossRef] [Green Version]
- Carmignotto, G.P.; Azzoni, A.R. On the expression of recombinant Cas9 protein in E. coli BL21(DE3) and BL21(DE3) Rosetta strains. J. Biotechnol. 2019, 306, 62–70. [Google Scholar] [CrossRef] [PubMed]
- D’Astolfo, D.S.; Pagliero, R.J.; Pras, A.; Karthaus, W.R.; Clevers, H.; Prasad, V.; Lebbink, R.J.; Rehmann, H.; Geijsen, N. Efficient Intracellular Delivery of Native Proteins. Cell 2015, 161, 674–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Yoon, J.; Kwon, D.; Han, M.-J.; Choi, S.; Park, S.; Lee, J.; Lee, K.; Lee, J.; Lee, S.; et al. Enhanced genome editing efficiency of CRISPR PLUS: Cas9 chimeric fusion proteins. Sci. Rep. 2021, 11, 16199. [Google Scholar] [CrossRef]
- Chaverra-Rodriguez, D.; Macias, V.M.; Hughes, G.L.; Pujhari, S.; Suzuki, Y.; Peterson, D.R.; Kim, D.; McKeand, S.; Rasgon, J.L. Targeted delivery of CRISPR-Cas9 ribonucleoprotein into arthropod ovaries for heritable germline gene editing. Nat. Commun. 2018, 9, 3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, A.; Matsumoto, D.; Kato, Y.; Honda, Y.; Morikawa, M.; Iwata, F.; Kobayashi, T.; Nakamura, C. Direct Delivery of Cas9-sgRNA Ribonucleoproteins into Cells Using a Nanoneedle Array. Appl. Sci. 2019, 9, 965. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766. [Google Scholar] [CrossRef]
- Yan, Q.-L.; Kong, H.-T.; Xia, K.; Zhang, Y.; Aladlbahi, A.; Shi, J.-Y.; Wang, L.-H.; Fan, C.-H.; Zhao, Y.; Zhu, Y. Expression and radiolabeling of Cas9 protein. Nucl. Sci. Tech. 2017, 28, 11. [Google Scholar] [CrossRef]
- Liu, J.; Gaj, T.; Yang, Y.; Wang, N.; Shui, S.; Kim, S.; Kanchiswamy, C.N.; Kim, J.-S.; Barbas, C.F., 3rd. Efficient delivery of nuclease proteins for genome editing in human stem cells and primary cells. Nat. Protoc. 2015, 10, 1842–1859. [Google Scholar] [CrossRef]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Akhmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186, Erratum in PLoS ONE 2014, 9, e106396. [Google Scholar] [CrossRef]



| Activity | Step | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 |
|---|---|---|---|---|---|---|---|
| Bacterial transformation | 1 | ON | |||||
| Glycerol preparation | 1 | 8 h | |||||
| Preparation of starting plate | 2 | ON | |||||
| Bacterial growth | 3–5 | 3–4 h | |||||
| Induction | 5 | ON | |||||
| Harvest | 6–7 | 1 h | |||||
| Lysis and clarification | 8–12 | 2.5 h | |||||
| IMAC | 13 | 1.5 h | |||||
| Buffer exchange | 14 | 30 min | |||||
| IEX | 16 | 1.5 h | |||||
| Buffer exchange | 17 | 30 min | |||||
| SDS-PAGE | 19 | 1.5 h | |||||
| Storage | 20–21 | 30 min |
| Volume (μL) | |
|---|---|
| H2O DEPC | To 50 μL |
| Buffer 10X Cas9 + DTT | 5 μL |
| His6−MBP−SpCas9 NLS−GFP | X μL (4 μg) |
| SgRNA | X μL (1120 ng) |
| Step | Suggested Procedure | Alternative |
|---|---|---|
| 3.2.2 IMAC | IMAC HiTrap FastFlow column (GE, Cat. no.: 17092104) connected to ÄKTA FPLC system (GE, Cat. no.: 29-0598-78 AB) | A. Perform IMAC with the column connected to a peristaltic pump. Follow absorbance at 280 nm using a spectrophotometer. Equilibrate the IMAC column with 10 CV on IMAC buffer A and measure the buffer absorbance before loading your protein sample. Make sure that this value is reached again after extensively washing bound protein. B. Perform batch incubation with IMAC resin. Follow the absorbance at 280 nm using a spectrophotometer. Equilibrate the resin in IMAC buffer A and measure the buffer absorbance before loading your protein sample. Make sure that this value is reached again after extensively washing bound protein. |
| 3.2.3 Buffer exchange | Perform buffer exchange in PD-10 column (GE, Cat. 17-0851-01) into IEX buffer A. | A. Buffer exchange the protein using centrifugal filter units with a cutoff >50 kDa and perform several dilution/concentration steps. Use IEX buffer A as the diluting solution. |
| 3.2.4 IEX | Cation exchange SpHp HighTrap column (GE, Cat. GE29-0513-24) connected to ÄKTA FPLC system (GE, Cat. no.: 29-0598-78 AB) | A. Perform IEX with the column connected to a peristaltic pump. Follow the absorbance at 280 nm using a spectrophotometer. Equilibrate the IMAC column with 10 CV on IEX buffer A and measure the buffer absorbance before loading your protein sample. Make sure that this value is reached again after extensively washing bound protein. Perform elution in a single step with 1 M NaCl concentration or in several steps with increasing NaCl concentrations. |
| 3.2.5 Buffer exchange | Perform buffer exchange in the PD-10 column (GE, Cat. 17-0851-01) into storage buffer. | A. Buffer exchange the protein using centrifugal filter units with a cutoff >50 kDa and perform several dilution/concentration steps. Use storage buffer as the diluting solution. B. Use a Superdex 200 16/600 size exclusion column (GE Healthcare), equilibrated in storage buffer for the final polishing step. |
| Problem | Possible Explanation | Solution |
|---|---|---|
| No protein expression | Plasmid loss | Retransform BL21 Rosetta or use another aliquot from your bacterial stock. Check plasmid presence in your bacterial stock. |
| High protein contamination during IMAC | Incomplete wash of nonspecific proteins | Do not elute protein until buffer absorbance from the washing fraction reaches the starting value. Try performing a washing step with IMAC buffer A containing 5 mM imidazole. This can lead to some Cas9 washing as well. |
| No protein binding to IEX column | Nucleic acid contamination. | Treat the protein fraction with benzonase. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fleitas, A.L.; Señorale, M.; Vidal, S. A Robust Expression and Purification Method for Production of SpCas9-GFP-MBP Fusion Protein for In Vitro Applications. Methods Protoc. 2022, 5, 44. https://doi.org/10.3390/mps5030044
Fleitas AL, Señorale M, Vidal S. A Robust Expression and Purification Method for Production of SpCas9-GFP-MBP Fusion Protein for In Vitro Applications. Methods and Protocols. 2022; 5(3):44. https://doi.org/10.3390/mps5030044
Chicago/Turabian StyleFleitas, Andrea Luciana, Mario Señorale, and Sabina Vidal. 2022. "A Robust Expression and Purification Method for Production of SpCas9-GFP-MBP Fusion Protein for In Vitro Applications" Methods and Protocols 5, no. 3: 44. https://doi.org/10.3390/mps5030044
APA StyleFleitas, A. L., Señorale, M., & Vidal, S. (2022). A Robust Expression and Purification Method for Production of SpCas9-GFP-MBP Fusion Protein for In Vitro Applications. Methods and Protocols, 5(3), 44. https://doi.org/10.3390/mps5030044