
Automated Dashboards for the Identification of Pathogenic Circulating Tumor DNA Mutations in Longitudinal Blood Draws of Cancer Patients


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
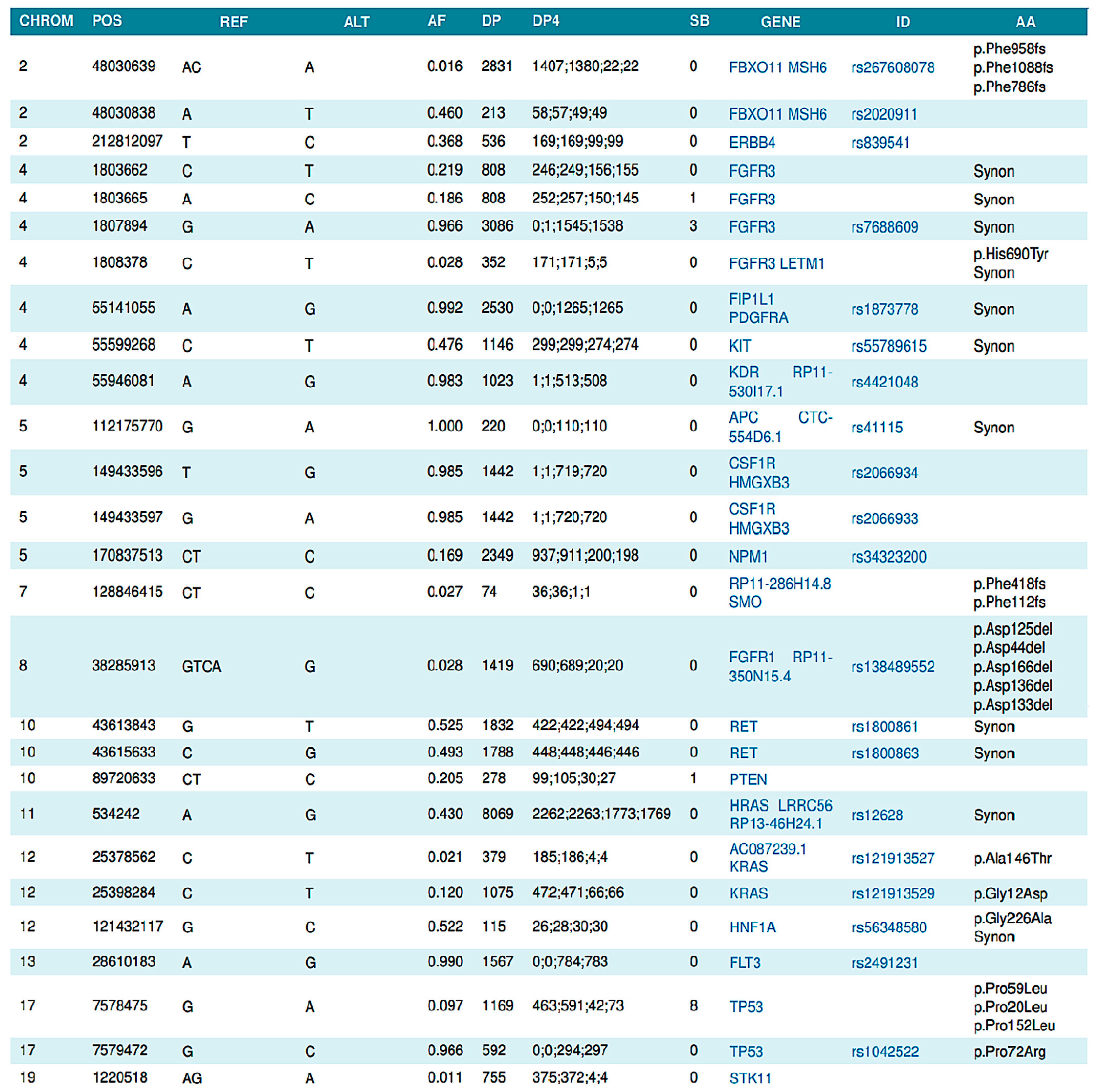
2.1. Genetic Sequencing Method
2.1.1. Circulating (Cell-Free) Tumor DNA Extraction
2.1.2. Next-Generation Sequencing
2.1.3. Data Analysis
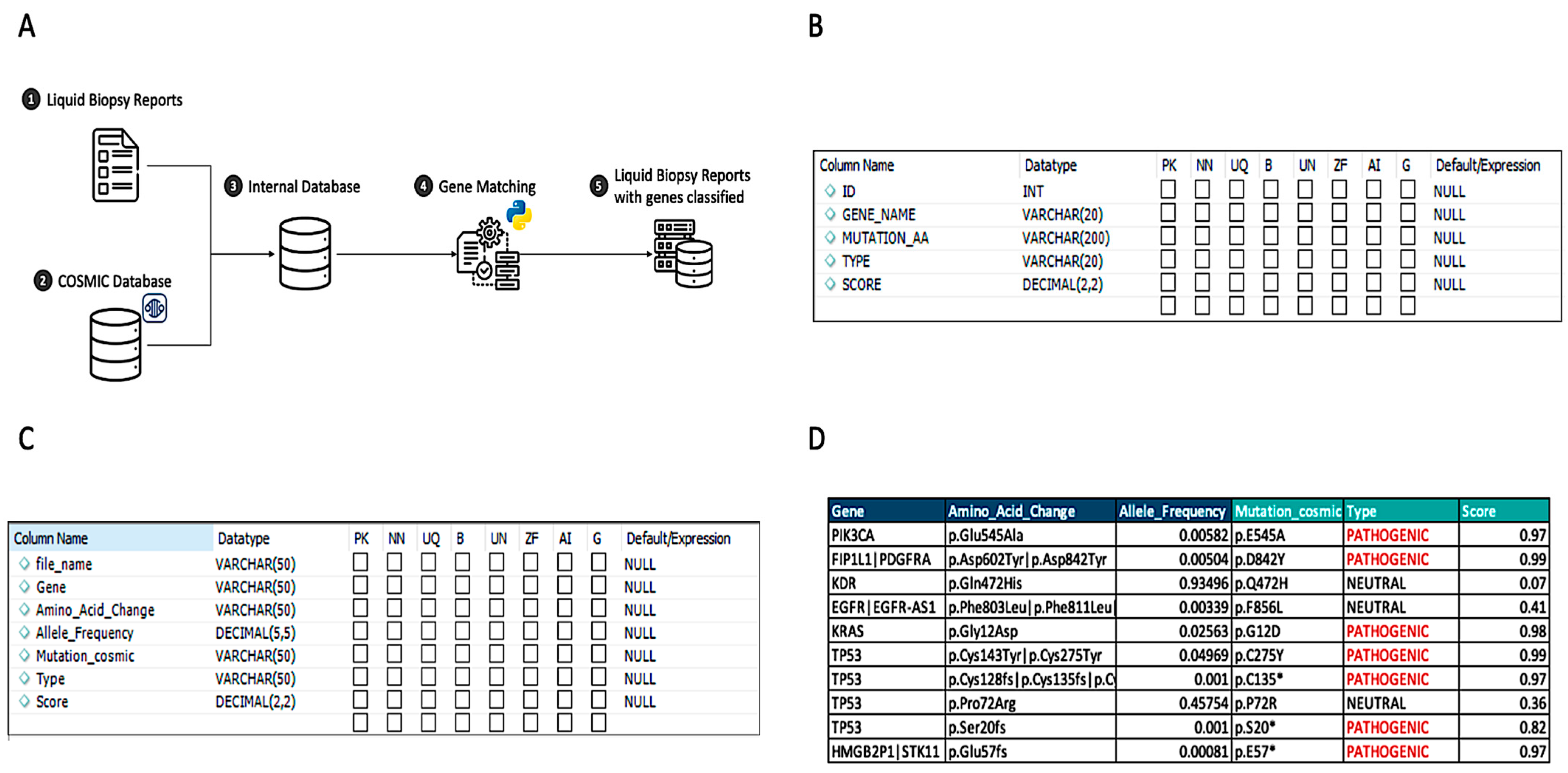
2.2. Pathogenic Matching Approach
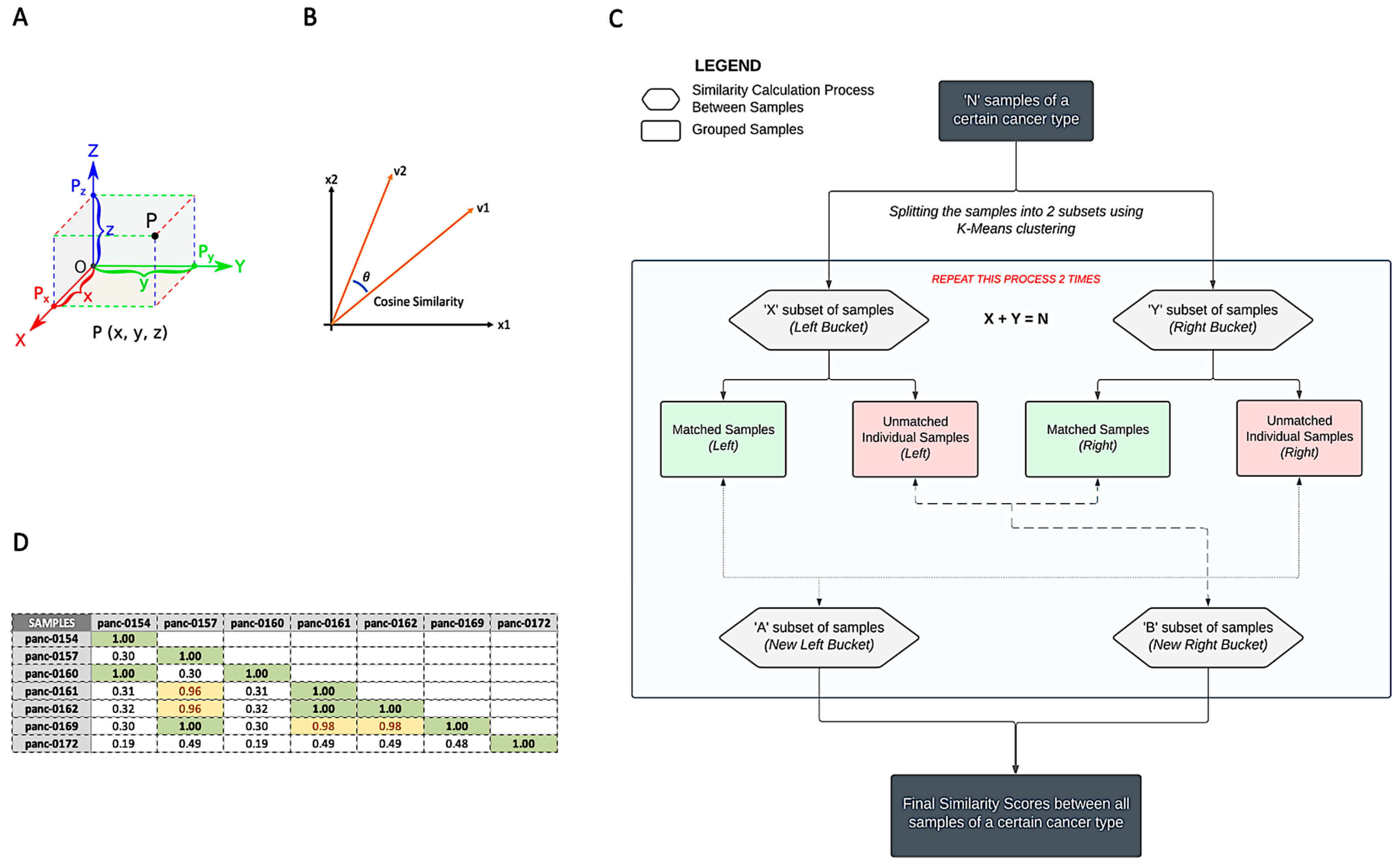
2.3. Patient Similarity Analysis
2.4. Longitudinal Data Visualization
3. Results
3.1. Pathogenic Mutation Matching
3.2. Patient Matching
3.3. Longitudinal Visualization
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Jian, D.; Sidorov, M.; Woo, R.W.L.; Kim, A.; Stone, D.E.; Nazarian, A.; Nosrati, M.; Ice, R.J.; de Semir, D.; et al. Pitfalls and Rewards of Setting Up a Liquid Biopsy Approach for the Detection of Driver Mutations in Circulating Tumor DNAs: Our Institutional Experience. J. Pers. Med. 2022, 12, 1845. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.-J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.-F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Crown, J. Use of Circulating Tumour DNA (ctDNA) for Measurement of Therapy Predictive Biomarkers in Patients with Cancer. J. Pers. Med. 2022, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Attard, G.; Bidard, F.-C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.; Montagut, C.; et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.K.; Park, B.H. Circulating tumor DNA: Current challenges for clinical utility. J. Clin. Investig. 2022, 132, e154941. [Google Scholar] [CrossRef] [PubMed]
- Levatić, J.; Salvadores, M.; Fuster-Tormo, F.; Supek, F. Mutational signatures are markers of drug sensitivity of cancer cells. Nat. Commun. 2022, 13, 2926. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, M.; Xu, S.; Huang, Z.; Grant, P.W. The Unsupervised Feature Selection Algorithms Based on Standard Deviation and Cosine Similarity for Genomic Data Analysis. Front. Genet. 2021, 12, 684100. [Google Scholar] [CrossRef] [PubMed]
- Strachna, O.; Cohen, M.A.; Ba, M.M.A.; Pfister, D.G.; Lee, N.Y.; Wong, R.J.; McBride, S.M.; Ba, R.R.M.; Kemeny, E.; Polubriaginof, F.C.G.; et al. Case study of the integration of electronic patient-reported outcomes as standard of care in a head and neck oncology practice: Obstacles and opportunities. Cancer 2020, 127, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Su, L.; Qian, C. Circulating tumor DNA: A promising biomarker in the liquid biopsy of cancer. Oncotarget 2016, 7, 48832–48841. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.A.; Velculescu, V.E. Blood-Based Analyses of Cancer: Circulating Tumor Cells and Circulating Tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udalov, A.; Kumar, L.; Gaudette, A.N.; Zhang, R.; Salomao, J.; Saigal, S.; Nosrati, M.; McAllister, S.D.; Desprez, P.-Y. Automated Dashboards for the Identification of Pathogenic Circulating Tumor DNA Mutations in Longitudinal Blood Draws of Cancer Patients. Methods Protoc. 2023, 6, 46. https://doi.org/10.3390/mps6030046
Udalov A, Kumar L, Gaudette AN, Zhang R, Salomao J, Saigal S, Nosrati M, McAllister SD, Desprez P-Y. Automated Dashboards for the Identification of Pathogenic Circulating Tumor DNA Mutations in Longitudinal Blood Draws of Cancer Patients. Methods and Protocols. 2023; 6(3):46. https://doi.org/10.3390/mps6030046
Chicago/Turabian StyleUdalov, Aleksandr, Lexman Kumar, Anna N. Gaudette, Ran Zhang, Joao Salomao, Sanjay Saigal, Mehdi Nosrati, Sean D. McAllister, and Pierre-Yves Desprez. 2023. "Automated Dashboards for the Identification of Pathogenic Circulating Tumor DNA Mutations in Longitudinal Blood Draws of Cancer Patients" Methods and Protocols 6, no. 3: 46. https://doi.org/10.3390/mps6030046
APA StyleUdalov, A., Kumar, L., Gaudette, A. N., Zhang, R., Salomao, J., Saigal, S., Nosrati, M., McAllister, S. D., & Desprez, P. -Y. (2023). Automated Dashboards for the Identification of Pathogenic Circulating Tumor DNA Mutations in Longitudinal Blood Draws of Cancer Patients. Methods and Protocols, 6(3), 46. https://doi.org/10.3390/mps6030046