New Therapies of Neovascular AMD—Beyond Anti-VEGFs


Abstract
:1. Introduction
2. Current Treatment Modalities of nAMD
2.1. Anti-VEGF Injections
2.2. Photodynamic Therapy
3. Rationale for Developing New Therapies
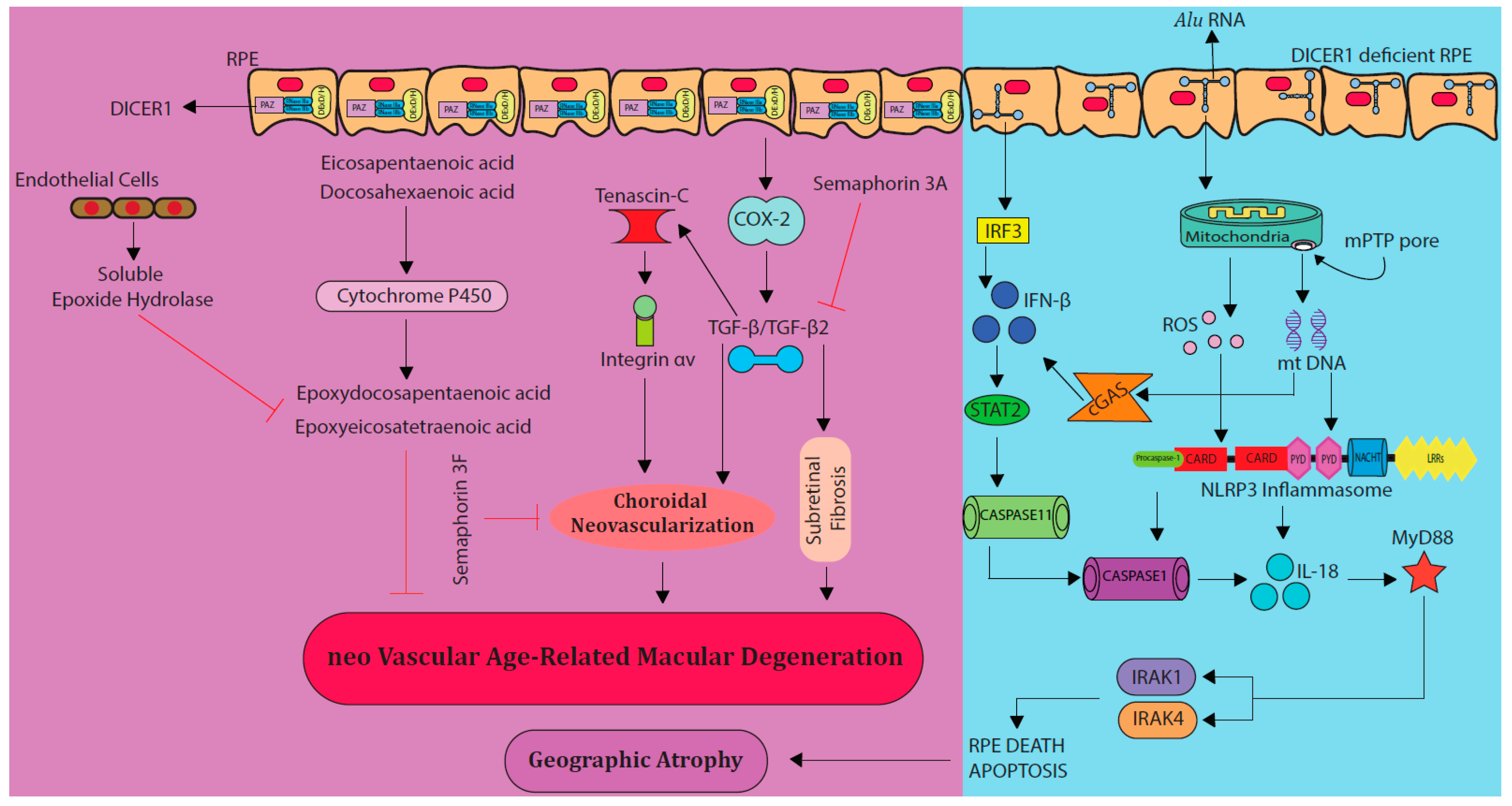
4. New Therapies for nAMD—Thinking beyond Anti-VEGFs
4.1. Semaphorin 3F
4.2. Tissue Factor Inhibition
4.3. Targeting the Cytochrome P450 Monooxygenase Pathway
4.4. Interleukin-33 Therapy
4.5. Targeted Intraceptor Nanoparticle Therapy
4.6. Targeting MyD88 Pathway and DICER 1
4.7. Interferon-β Therapy
4.8. Intravenous Injection of Immune Globulin
4.9. Inhibitors of Integrins
4.10. COX-2 Inhibitors
4.11. Inhibition of CCR3
5. Targeting Other Signaling Pathways with Involvement of VEGF
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Augood, C.A.; Vingerling, J.R.; de Jong, P.T.; Chakravarthy, U.; Seland, J.; Soubrane, G.; Tomazzoli, L.; Topouzis, F.; Bentham, G.; Rahu, M.; et al. Prevalence of age-related maculopathy in older Europeans: The European Eye Study (EUREYE). Arch. Ophthalmol. 2006, 124, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34. [Google Scholar] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Ferris, F.L.; Wilkinson, C.P.; Bird, A.; Chakravarthy, U.; Chew, E.; Csaky, K.; Sadda, S.R. Clinical Classification of Age-related Macular Degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Kolar, P. Classification and clinical features of AMD. In Age-Related Macular Degeneration-Etiology, Diagnosis and Management—A Glance at the Future; InTech: London, UK, 2013; pp. 105–132. [Google Scholar]
- Nowak, J.Z. Age-related macular degeneration (AMD): Pathogenesis and therapy. Pharmacol. Rep. 2006, 58, 353–363. [Google Scholar] [PubMed]
- Wang, H.; Hartnett, M.E. Regulation of signaling events involved in the pathophysiology of neovascular AMD. Mol. Vis. 2016, 22, 189–202. [Google Scholar] [PubMed]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [PubMed] [Green Version]
- Amadio, M.; Govoni, S.; Pascale, A. Targeting VEGF in eye neovascularization: What’s new? A comprehensive review on current therapies and oligonucleotide-based interventions under development. Pharmacol. Res. 2016, 103, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, M.; Marin-Castano, M.E. Cigarette smoke-related hydroquinone dysregulates MCP-1, VEGF and PEDF expression in retinal pigment epithelium in vitro and in vivo. PLoS ONE 2011, 6, e16722. [Google Scholar] [CrossRef] [PubMed]
- Cherepanoff, S.; McMenamin, P.; Gillies, M.C.; Kettle, E.; Sarks, S.H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 2010, 94, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Raoul, W.; Auvynet, C.; Camelo, S.; Guillonneau, X.; Feumi, C.; Combadiere, C.; Sennlaub, F. CCL2/CCR2 and CX3CL1/CX3CR1 chemokine axes and their possible involvement in age-related macular degeneration. J. Neuroinflamm. 2010, 7, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telander, D.G. Inflammation and age-related macular degeneration (AMD). Semin. Ophthalmol. 2011, 26, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Schlottmann, P.G.; Alezzandrini, A.A.; Zas, M.; Rodriguez, F.J.; Luna, J.D.; Wu, L. New treatment modalities for neovascular age-related macular degeneration. Asia Pac. J. Ophthalmol. 2017, 6, 514–519. [Google Scholar]
- Lai, K.; Landa, G. Current choice of treatments for neovascular AMD. Expert Rev. Clin. Pharmacol. 2015, 8, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Villegas, V.M.; Aranguren, L.A.; Kovach, J.L.; Schwartz, S.G.; Flynn, H.W. Current advances in the treatment of neovascular age-related macular degeneration. Expert Opin. Drug Deliv. 2017, 14, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, J.; Fei, D.; Beyer, J.C.; Ryan, A.; Rangell, L.; Shiu, V.; Damico, L.A. Pharmacokinetics and retinal distribution of ranibizumab, a humanized antibody fragment directed against VEGF-A, following intravitreal administration in rabbits. Retina 2007, 27, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Krohne, T.U.; Eter, N.; Holz, F.G.; Meyer, C.H. Intraocular pharmacokinetics of bevacizumab after a single intravitreal injection in humans. Am. J. Ophthalmol. 2008, 146, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D.; et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef] [PubMed]
- Dugel, P.U.; Jaffe, G.J.; Sallstig, P.; Warburton, J.; Weichselberger, A.; Wieland, M.; Singerman, L. Brolucizumab Versus Aflibercept in Participants with Neovascular Age-Related Macular Degeneration: A Randomized Trial. Ophthalmology 2017, 124, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- DeCroos, F.C.; Reed, D.; Adam, M.K.; Salz, D.; Gupta, O.P.; Ho, A.C.; Regillo, C.D. Treat-and-Extend Therapy Using Aflibercept for Neovascular Age-related Macular Degeneration: A Prospective Clinical Trial. Am. J. Ophthalmol. 2017, 180, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Souied, E.H.; Devin, F.; Mauget-Faysse, M.; Kolar, P.; Wolf-Schnurrbusch, U.; Framme, C.; Gaucher, D.; Querques, G.; Stumpp, M.T.; Wolf, S. Treatment of exudative age-related macular degeneration with a designed ankyrin repeat protein that binds vascular endothelial growth factor: A phase I/II study. Am. J. Ophthalmol. 2014, 158, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthy, U.; Bailey, C.; Brown, D.; Campochiaro, P.; Chittum, M.; Csaky, K.; Tufail, A.; Yates, P.; Cech, P.; Giraudon, M.; et al. Phase I Trial of Anti–Vascular Endothelial Growth Factor/Anti-angiopoietin 2 Bispecific Antibody RG7716 for Neovascular Age-Related Macular Degeneration. Ophthalmol. Retina 2017, 1, 474–485. [Google Scholar] [CrossRef]
- Brown, D.M. Phase 1 study of combination therapy with nesvacumab and aflibercept for neovascular AMD and diabetic macular edema. In Proceedings of the American Academy of Ophthalmology Annual Meeting, Chicago, IL, USA, 14–18 October 2016. [Google Scholar]
- Van den Bergh, H. Photodynamic therapy of age-related macular degeneration: History and principles. Semin. Ophthalmol. 2001, 16, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Bressler, N.M.; Chang, T.S.; Fine, J.T.; Dolan, C.M.; Ward, J. Improved vision-related function after ranibizumab vs photodynamic therapy: A randomized clinical trial. Arch. Ophthalmol. 2009, 127, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Daniel, E.; Toth, C.A.; Grunwald, J.E.; Jaffe, G.J.; Martin, D.F.; Fine, S.L.; Huang, J.; Ying, G.S.; Hagstrom, S.A.; Winter, K.; et al. Risk of scar in the comparison of age-related macular degeneration treatments trials. Ophthalmology 2014, 121, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, J.E.; Pistilli, M.; Daniel, E.; Ying, G.S.; Pan, W.; Jaffe, G.J.; Toth, C.A.; Hagstrom, S.A.; Maguire, M.G.; Martin, D.F. Incidence and Growth of Geographic Atrophy during 5 Years of Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 2017, 124, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, S.; Acquah, K.; Mruthyunjaya, P.; Grossman, D.S.; Lee, P.P.; Sloan, F.A. Ocular complications after anti-vascular endothelial growth factor therapy in Medicare patients with age-related macular degeneration. Am. J. Ophthalmol. 2011, 152, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Johnson, D.; Abouammoh, M.; Hollands, S.; Brissette, A. Rate of serious adverse effects in a series of bevacizumab and ranibizumab injections. Can. J. Ophthalmol. 2012, 47, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Pershing, S.; Bakri, S.J.; Moshfeghi, D.M. Ocular hypertension and intraocular pressure asymmetry after intravitreal injection of anti-vascular endothelial growth factor agents. Ophthalmic Surg. Lasers Imaging Retina 2013, 44, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Gregori, N.Z.; Flynn, H.W., Jr.; Schwartz, S.G.; Rosenfeld, P.J.; Vaziri, K.; Moshfeghi, A.A.; Fortun, J.A.; Kovach, J.L.; Dubovy, S.R.; Albini, T.A.; et al. Current Infectious Endophthalmitis Rates After Intravitreal Injections of Anti-Vascular Endothelial Growth Factor Agents and Outcomes of Treatment. Ophthalmic Surg. Lasers Imaging Retina 2015, 46, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: A multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299. [Google Scholar] [CrossRef] [PubMed]
- Gaur, P.; Bielenberg, D.R.; Samuel, S.; Bose, D.; Zhou, Y.; Gray, M.J.; Dallas, N.A.; Fan, F.; Xia, L.; Lu, J.; et al. Role of class 3 semaphorins and their receptors in tumor growth and angiogenesis. Clin. Cancer Res. 2009, 15, 6763–6770. [Google Scholar] [CrossRef] [PubMed]
- Buehler, A.; Sitaras, N.; Favret, S.; Bucher, F.; Berger, S.; Pielen, A.; Joyal, J.-S.; Juan, A.M.; Martin, G.; Schlunck, G.; et al. Semaphorin 3F forms an anti-angiogenic barrier in outer retina. FEBS Lett. 2013, 587, 1650–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liegl, R.; Gong, Y.; Bühler, A.; Cakir, B.; Meng, S.S.; Burnim, S.B.; Liu, C.-H.; Reuer, T.; Zhang, P.; et al. Sema3f Protects Against Subretinal Neovascularization In Vivo. EBioMedicine 2017, 18, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. The role of tissue factor and factor VIIa in hemostasis. Anesth. Anal. 2009, 108, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Guo, B.; Li, Y.; Zhu, B. Tissue factor in tumor microenvironment: A systematic review. J. Hematol. Oncol. 2014, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.; Cao, X.; Shen, D.; Tuo, J.; Parver, L.M.; Rickles, F.R.; Chan, C.C. Evidence for enhanced tissue factor expression in age-related macular degeneration. Lab. Investig. 2011, 91, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-F.; Zou, X.-L. Tissue factor with age-related macular degeneration. Int. J. Ophthalmol. 2012, 5, 609–613. [Google Scholar] [PubMed]
- Grossniklaus, H.E.; Ling, J.X.; Wallace, T.M.; Dithmar, S.; Lawson, D.H.; Cohen, C.; Elner, V.M.; Elner, S.G.; Sternberg, P., Jr. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol. Vis. 2002, 8, 119–126. [Google Scholar] [PubMed]
- Wang, L.; Yang, Z.; Yu, Y.; Cui, C.; Guan, H.; Chen, H. Blockage of tissue factor ameliorates the lesion of laser-induced choroidal neovascularization in mice. Exp. Eye Res. 2014, 127, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Gian Marco Tosi, M.B.; Maurizio, O.; Federico, G. New molecular targets for the treatment of neovascular age-related macular degeneration. Trans. Med. Rep. 2017, 1, 96–104. [Google Scholar]
- Arnold, C.; Konkel, A.; Fischer, R.; Schunck, W.H. Cytochrome P450-dependent metabolism of omega-6 and omega-3 long-chain polyunsaturated fatty acids. Pharmacol. Rep. 2010, 62, 536–547. [Google Scholar] [CrossRef]
- Yanai, R.; Mulki, L.; Hasegawa, E.; Takeuchi, K.; Sweigard, H.; Suzuki, J.; Gaissert, P.; Vavvas, D.G.; Sonoda, K.H.; Rothe, M.; et al. Cytochrome P450-generated metabolites derived from omega-3 fatty acids attenuate neovascularization. Proc. Natl. Acad. Sci. USA 2014, 111, 9603–9608. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, E.; Inafuku, S.; Mulki, L.; Okunuki, Y.; Yanai, R.; Smith, K.E.; Kim, C.B.; Klokman, G.; Bielenberg, D.R.; Puli, N.; et al. Cytochrome P450 monooxygenase lipid metabolites are significant second messengers in the resolution of choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2017, 114, e7545–e7553. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, E.; Taguchi, H.; Anand, A.; Ambati, B.K.; Gragoudas, E.S.; Miller, J.W.; Adamis, A.P.; Ambati, J. Targeted Disruption of the CD18 or ICAM-1 Gene Inhibits Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2743–2749. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Liegl, R.; Wang, Z.; Gong, Y.; Liu, C.H.; Sun, Y.; Cakir, B.; Burnim, S.B.; Meng, S.S.; Lofqvist, C.; et al. Adiponectin Mediates Dietary Omega-3 Long-Chain Polyunsaturated Fatty Acid Protection against Choroidal Neovascularization in Mice. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3862–3870. [Google Scholar] [CrossRef] [PubMed]
- Chew, Y.E.; Clemons, T.E.; SanGiovanni, J.P.; Danis, R.; Ferris, F.L.; Elman, M.; Antoszyk, A.; Ruby, A.; Orth, D.; Bressler, S. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: The Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA 2013, 309, 2005–2015. [Google Scholar]
- Souied, E.H.; Aslam, T.; Garcia-Layana, A.; Holz, F.G.; Leys, A.; Silva, R.; Delcourt, C. Omega-3 fatty acids and age-related macular degeneration. Ophthalmic Res. 2015, 55, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Merle, B.M.; Benlian, P.; Puche, N.; Bassols, A.; Delcourt, C.; Souied, E.H. Circulating omega-3 fatty acids and neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, A.A.; Oldham, E.R.; Murphy, E.E.; Schmitz, J.; Pflanz, S.; Kastelein, R.A. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J. Immunol. 2007, 179, 2551–2555. [Google Scholar] [CrossRef] [PubMed]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Barbour, M.; Allan, D.; Xu, H.; Pei, C.; Chen, M.; Niedbala, W.; Fukada, S.Y.; Besnard, A.-G.; Alves-Filho, J.C.; Tong, X.; et al. IL-33 attenuates the development of experimental autoimmune uveitis. Eur. J. Immunol. 2014, 44, 3320–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoropoulou, S.; Copland, D.A.; Liu, J.; Wu, J.; Gardner, P.J.; Ozaki, E.; Doyle, S.L.; Campbell, M.; Dick, A.D. Interleukin-33 regulates tissue remodelling and inhibits angiogenesis in the eye. J. Pathol. 2017, 241, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, X.; Hirano, Y.; Tyagi, P.; Barabás, P.; Uehara, H.; Miya, T.R.; Singh, N.; Archer, B.; Qazi, Y.; et al. Targeted Intraceptor Nanoparticle Therapy Reduces Angiogenesis and Fibrosis in Primate and Murine Macular Degeneration. ACS Nano 2013, 7, 3264–3275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Bohner, A.; Bhuvanagiri, S.; Uehara, H.; Upadhyay, A.K.; Emerson, L.L.; Bondalapati, S.; Muddana, S.K.; Fang, D.; Li, M.; et al. Targeted Intraceptor Nanoparticle for Neovascular Macular Degeneration: Preclinical Dose Optimization and Toxicology Assessment. Mol. Ther. 2017, 25, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Schutze, C.; Wedl, M.; Baumann, B.; Pircher, M.; Hitzenberger, C.K.; Schmidt-Erfurth, U. Progression of retinal pigment epithelial atrophy in antiangiogenic therapy of neovascular age-related macular degeneration. Am. J. Ophthalmol. 2015, 159, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Bhisitkul, R.B.; Mendes, T.S.; Rofagha, S.; Enanoria, W.; Boyer, D.S.; Sadda, S.R.; Zhang, K. Macular atrophy progression and 7-year vision outcomes in subjects from the ANCHOR, MARINA, and HORIZON studies: The SEVEN-UP study. Am. J. Ophthalmol. 2015, 159, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Enslow, R.; Bhuvanagiri, S.; Vegunta, S.; Cutler, B.; Neff, M.; Stagg, B. Association of anti-VEGF injections with progression of geographic atrophy. Ophthalmol. Eye Dis. 2016, 8, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Fine, S.L.; Ying, G.S.; Jaffe, G.J.; Grunwald, J.E.; Toth, C.; Redford, M.; Ferris, F.L., 3rd. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: Two-year results. Ophthalmology 2012, 119, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; Yang, L. ALU ternative Regulation for Gene Expression. Trends Cell Biol. 2017, 27, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef] [PubMed]
- Yerramothu, P.; Vijay, A.K.; Willcox, M.D.P. Inflammasomes, the eye and anti-inflammasome therapy. Eye 2017, 32, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Fukuda, S.; Banerjee, D.; Kim, Y.; Fu, D.; Apicella, I.; Varshney, A.; Yasuma, R.; Fowler, B.J.; Baghdasaryan, E.; et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 2017, 24, 50. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, E.; Anand, A.; Ambati, B.K.; van Rooijen, N.; Ambati, J. Macrophage Depletion Inhibits Experimental Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3578–3585. [Google Scholar] [CrossRef] [Green Version]
- Levy, O.; Calippe, B.; Lavalette, S.; Hu, S.J.; Raoul, W.; Dominguez, E.; Housset, M.; Paques, M.; Sahel, J.A.; Bemelmans, A.P.; et al. Apolipoprotein E promotes subretinal mononuclear phagocyte survival and chronic inflammation in age-related macular degeneration. EMBO Mol. Med. 2015, 7, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa-Heidmann, D.G.; Suner, I.J.; Hernandez, E.P.; Monroy, D.; Csaky, K.G.; Cousins, S.W. Macrophage Depletion Diminishes Lesion Size and Severity in Experimental Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3586–3592. [Google Scholar] [CrossRef] [Green Version]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Schmidt, H.; Mildner, A.; Knobeloch, K.P.; Hanisch, U.K.; Raasch, J.; Merkler, D.; Detje, C.; Gutcher, I.; Mages, J.; et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 2008, 28, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Teige, I.; Treschow, A.; Teige, A.; Mattsson, R.; Navikas, V.; Leanderson, T.; Holmdahl, R.; Issazadeh-Navikas, S. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J. Immunol. 2003, 170, 4776–4784. [Google Scholar] [CrossRef] [PubMed]
- Luckoff, A.; Caramoy, A.; Scholz, R.; Prinz, M.; Kalinke, U.; Langmann, T. Interferon-beta signaling in retinal mononuclear phagocytes attenuates pathological neovascularization. EMBO Mol. Med. 2016, 8, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L.; Merrill, J.T.; McInnes, I.B.; Peakman, M. Optimization of current and future therapy for autoimmune diseases. Nat. Med. 2012, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Haji Abdolvahab, M.; Mofrad, M.R.; Schellekens, H. Interferon Beta: From Molecular Level to Therapeutic Effects. Int. Rev. Cell Mol. Biol. 2016, 326, 343–372. [Google Scholar] [PubMed]
- Sottini, A.; Capra, R.; Serana, F.; Chiarini, M.; Caimi, L.; Imberti, L. Interferon-beta therapy monitoring in multiple sclerosis patients. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Walther, E.U.; Hohlfeld, R. Multiple sclerosis: Side effects of interferon beta therapy and their management. Neurology 1999, 53, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Bayas, A.; Rieckmann, P. Managing the adverse effects of interferon-beta therapy in multiple sclerosis. Drug Saf. 2000, 22, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, E.W. Intravenous immune globulin in autoimmune and inflammatory diseases. N. Engl. J. Med. 2012, 367, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Ammann, A.J.; Ashman, R.F.; Buckley, R.H.; Hardie, W.R.; Krantmann, H.J.; Nelson, J.; Ochs, H.; Stiehm, E.R.; Tiller, T.; Wara, D.W.; et al. Use of intravenous gamma-globulin in antibody immunodeficiency: Results of a multicenter controlled trial. Clin. Immunol. Immunopathol. 1982, 22, 60–67. [Google Scholar] [CrossRef]
- Buckley, R.H.; Schiff, R.I. The Use of Intravenous Immune Globulin in Immunodeficiency Diseases. N. Engl. J. Med. 1991, 325, 110–117. [Google Scholar] [PubMed]
- Imbach, P.; Barandun, S.; d’Apuzzo, V.; Baumgartner, C.; Hirt, A.; Morell, A.; Rossi, E.; Schoni, M.; Vest, M.; Wagner, H.P. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet 1981, 1, 1228–1231. [Google Scholar] [CrossRef]
- Bogdanovich, S.; Kim, Y.; Mizutani, T.; Yasuma, R.; Tudisco, L.; Cicatiello, V.; Bastos-Carvalho, A.; Kerur, N.; Hirano, Y.; Baffi, J.Z.; et al. Human IgG1 antibodies suppress angiogenesis in a target-independent manner. Signal Transduct. Target Ther. 2016, 1, 15001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuma, R.; Cicatiello, V.; Mizutani, T.; Tudisco, L.; Kim, Y.; Tarallo, V.; Bogdanovich, S.; Hirano, Y.; Kerur, N.; Li, S.; et al. Intravenous immune globulin suppresses angiogenesis in mice and humans. Signal Transduct. Target. Ther. 2016, 1, 15002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danen, E.H.J. Integrins: An Overview of Structural and Functional Aspects; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Nandrot, E.F.; Kim, Y.; Brodie, S.E.; Huang, X.; Sheppard, D.; Finnemann, S.C. Loss of Synchronized Retinal Phagocytosis and Age-related Blindness in Mice Lacking αvβ5 Integrin. J. Exp. Med. 2004, 200, 1539–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallavarapu, M.; Finnemann, S.C. Neural retina and MerTK-independent apical polarity of alphavbeta5 integrin receptors in the retinal pigment epithelium. Adv. Exp. Med. Biol. 2010, 664, 123–131. [Google Scholar] [PubMed]
- Kaiser, P.; Boyer, D.; Campochiaro, P.; Guerrero-Naranjo, J.L.; Heier, J.; Kornfield, J.; Kuppermann, B.; Quiroz-Mercado, H.; Salinas Longoria, S.; Schwartz, S. Integrin Peptide Therapy: The First Wet AMD Experience. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2177. [Google Scholar]
- Santulli, R.J.; Kinney, W.A.; Ghosh, S.; Decorte, B.L.; Liu, L.; Tuman, R.W.; Zhou, Z.; Huebert, N.; Bursell, S.E.; Clermont, A.C.; et al. Studies with an orally bioavailable alpha V integrin antagonist in animal models of ocular vasculopathy: Retinal neovascularization in mice and retinal vascular permeability in diabetic rats. J. Pharmacol. Exp. Ther. 2008, 324, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Hoffmann, S.; Eichler, W.; Friedrichs, U.; Wang, Y.S.; Wiedemann, P. Inhibition of experimental choroidal neovascularization in rats by an alpha(v)-integrin antagonist. Curr. Eye Res. 2004, 28, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, F.; Lu, F.; Xu, S.; Hu, W.; Huang, J.; Gu, Q.; Sun, X. The antiangiogenic effects of integrin α5β1 inhibitor (ATN-161) in vitro and in vivo. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7213–7220. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.K.; Kociok, N.; Zahn, G.; Vossmeyer, D.; Stragies, R.; Muether, P.S.; Joussen, A.M. Modulation of hypoxia-induced neovascularization by JSM6427, an integrin alpha5beta1 inhibiting molecule. Curr. Eye Res. 2007, 32, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Kuppermann, B.D.; On Behalf of the Ophthotech Study Group. Inhibition of 5β1 Integrin in Neovascular AMD—A Phase 1 Study. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1252. [Google Scholar]
- Chiquet-Ehrismann, R.; Chiquet, M. Tenascins: Regulation and putative functions during pathological stress. J. Pathol. 2003, 200, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Nicolo, M.; Piccolino, F.C.; Zardi, L.; Giovannini, A.; Mariotti, C. Detection of tenascin-C in surgically excised choroidal neovascular membranes. Graefes Arch. Clin. Exp. Ophthalmol. 2000, 238, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Yoshida, S.; Zhou, Y.; Nakama, T.; Ishikawa, K.; Arima, M.; Nakao, S.; Sassa, Y.; Takeda, A.; Hisatomi, T.; et al. Tenascin-C promotes angiogenesis in fibrovascular membranes in eyes with proliferative diabetic retinopathy. Mol. Vis. 2016, 22, 436–445. [Google Scholar] [PubMed]
- Kobayashi, Y.; Yoshida, S.; Zhou, Y.; Nakama, T.; Ishikawa, K.; Kubo, Y.; Arima, M.; Nakao, S.; Hisatomi, T.; Ikeda, Y.; et al. Tenascin-C secreted by transdifferentiated retinal pigment epithelial cells promotes choroidal neovascularization via integrin alphaV. Lab. Investig. 2016, 96, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef] [PubMed]
- Connor, T.B.; Roberts, A.B.; Sporn, M.B.; Danielpour, D.; Dart, L.L.; Michels, R.G.; de Bustros, S.; Enger, C.; Kato, H.; Lansing, M. Correlation of fibrosis and transforming growth factor-beta type 2 levels in the eye. J. Clin. Investig. 1989, 83, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.; Puklin, J.E.; Frank, R.N. Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3178–3188. [Google Scholar]
- Zhang, R.; Liu, Z.; Zhang, H.; Zhang, Y.; Lin, D. The COX-2-Selective Antagonist (NS-398) Inhibits Choroidal Neovascularization and Subretinal Fibrosis. PLoS ONE 2016, 11, e0146808. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.I.; Ijzerman, A.P. Small molecule antagonists for chemokine CCR3 receptors. Med. Res. Rev. 2009, 30, 778–817. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Baffi, J.Z.; Kleinman, M.E.; Cho, W.G.; Nozaki, M.; Yamada, K.; Kaneko, H.; Albuquerque, R.J.; Dridi, S.; Saito, K.; et al. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature 2009, 460, 225–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Han, X.; Gambhir, D.; Becker, S.; Kunz, E.; Liu, A.J.; Hartnett, M.E. Retinal inhibition of CCR3 induces retinal cell death in a murine model of choroidal neovascularization. PLoS ONE 2016, 11, e0157748. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.H.; Kim, J.H.; Yang, W.; Kim, H.; Chang, S.; Kim, D.; Chang, M.; Lee, K.; Chung, J.; Kim, J.H. Anti-complement component 5 antibody targeting MG4 domain inhibits choroidal neovascularization. Oncotarget 2017, 8, 45506–45516. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Fujiu, K.; Manabe, I.; Nishida, J.; Yamagishi, R.; Nagai, R.; Yanagi, Y. Choroidal neovascularization is inhibited via an intraocular decrease of inflammatory cells in mice lacking complement component C3. Sci. Rep. 2015, 5, 15702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlin, B.; Schnabolk, G.; Joseph, K.; Raikwar, H.; Kunchithapautham, K.; Johnson, K.; Moore, K.; Wang, Y.; Rohrer, B. Connecting the innate and adaptive immune responses in mouse choroidal neovascularization via the anaphylatoxin C5a and γδT-cells. Sci. Rep. 2016, 6, 23794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrer, B.; Long, Q.; Coughlin, B.; Wilson, R.B.; Huang, Y.; Qiao, F.; Tang, P.H.; Kunchithapautham, K.; Gilkeson, G.S.; Tomlinson, S. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3056–3064. [Google Scholar] [CrossRef] [PubMed]
- Ntumba, K.; Akla, N.; Oh, S.P.; Eichmann, A.; Larrivée, B. BMP9/ALK1 inhibits neovascularization in mouse models of age-related macular degeneration. Oncotarget 2016, 7, 55957–55969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretz, C.A.; Divoky, V.; Prchal, J.; Kunz, E.; Simmons, A.B.; Wang, H.; Hartnett, M.E. Erythropoietin Signaling Increases Choroidal Macrophages and Cytokine Expression, and Exacerbates Choroidal Neovascularization. Sci. Rep. 2018, 8, 2161. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Li, K.R.; Li, X.M.; Yao, J.; Qin, J.; Yan, B. Long non-coding RNAs: New players in ocular neovascularization. Mol. Biol. Rep. 2014, 41, 4493–4505. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Robredo, P.; Selvam, S.; Powner, M.B.; Sim, D.A.; Fruttiger, M. Neuropilin 1 involvement in choroidal and retinal neovascularisation. PLoS ONE 2017, 12, e0169865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Takeda, A.; Yoshimura, T.; Oshima, Y.; Sonoda, K.-H.; Ishibashi, T. A Novel Platelet-Activating Factor Receptor Antagonist Inhibits Choroidal Neovascularization and Subretinal Fibrosis. PLoS ONE 2013, 8, e68173. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fowler, B.J.; Kim, Y.; Yasuma, R.; Krueger, L.A.; Gelfand, B.D.; Ambati, J. Nucleoside Reverse Transcriptase Inhibitors Suppress Laser-Induced Choroidal Neovascularization in Mice. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7122–7129. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Bailey, C.; Brown, D.; Campochiaro, P.; Chittum, M.; Csaky, K.; Tufail, A.; Yates, P.; Cech, P.; Giraudon, M.; et al. STAT3 Activation in Circulating Monocytes Contributes to Neovascular Age-Related Macular Degeneration. Curr. Mol. Med. 2016, 16, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, W.; Han, S.; Meng, Z.; Zhao, L.; Yin, Y.; Wang, Y.; Li, J. TGF-β participates choroid neovascularization through Smad2/3-VEGF/TNF-α signaling in mice with Laser-induced wet age-related macular degeneration. Sci. Rep. 2017, 7, 9672. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Shi, H.; Zhu, R.; Li, L.; Qin, B.; Kang, L.; Chen, H.; Guan, H. Inhibition of YAP ameliorates choroidal neovascularization via inhibiting endothelial cell proliferation. Mol. Vis. 2018, 24, 83–93. [Google Scholar] [PubMed]
- Lambert, N.G.; Zhang, X.; Rai, R.R.; Uehara, H.; Choi, S.; Carroll, L.S.; Das, S.K.; Cahoon, J.M.; Kirk, B.H.; Bentley, B.M.; et al. Subretinal AAV2.COMP-Ang1 suppresses choroidal neovascularization and vascular endothelial growth factor in a murine model of age-related macular degeneration. Exp. Eye Res. 2016, 145, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, F.; Matlock, G.; Chen, Q.; Zhou, K.; Du, Y.; Wang, X.; Ma, J.X. Therapeutic Effects of PPARalpha Agonist on Ocular Neovascularization in Models Recapitulating Neovascular Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5065–5075. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.D.; Schachar, R.A.; Nduaka, C.I.; Sperling, M.; Klamerus, K.J.; Chi-Burris, K.; Yan, E.; Paggiarino, D.A.; Rosenblatt, I.; Aitchison, R.; et al. Evaluation of the siRNA PF-04523655 versus ranibizumab for the treatment of neovascular age-related macular degeneration (MONET Study). Ophthalmology 2012, 119, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Kothary, P.C.; Badhwar, J.; Weng, C.; Del Monte, M.A. Impaired Intracellular Signaling May Allow Up-Regulation of CTGF-Synthesis and Secondary Peri-Retinal Fibrosis in Human Retinal Pigment Epithelial Cells from Patients with Age-Related Macular Degeneration. In Retinal Degenerative Diseases: Laboratory and Therapeutic Investigations; Anderson, R.E., Hollyfield, J.G., LaVail, M.M., Eds.; Springer: New York, NY, USA, 2010; pp. 419–428. [Google Scholar]


{kind=link}
| Signaling/Inhibitor | Key Molecules/Proteins Involved | Findings | Reference | |
|---|---|---|---|---|
| 1 | Complement pathway | C3a, C5a, monocyte chemoattractant protein-1 (MCP-1), VEGF, and MG4 domain, IL-17, γδ T-cells | Antibody-mediated neutralization of C3a, C5a, MG4 domains of β chain, or pharmacological inhibition of their receptors inhibited CNV in mouse nAMD | Jo et al., 2017 [109]; Nozaki et al., 2006 [12]; Tan et al., 2015 [110]; Coughlin et al., 2015 [111]; Robrer et al., 2009 [112] |
| 2 | BMP9/Alk1 signaling | BMP9, Alk1, VEGF, and VEGFR2 | Activating Alk1 signaling inhibited growth of blood vessels in nAMD mouse model | Ntumba et al., 2016 [113] |
| 3 | Erythropoietin signaling | Erythropoietin, macrophages, CCL2, CXCL10, CCL22, IL-6, and IL-10 | Increased erythropoietin signaling is associated with increased CNV in mice | Bretz et al., 2018 [114] |
| 4 | Long non-coding RNAs | MAPK signaling, Vax2osl, and Vax2os2 | 326 or 51 long non-coding RNAs that play a role in human nAMD were identified and their dysregulation could provide novel insights into nAMD treatments | Xu et al., 2014 [115] |
| 5 | Neuropilin 1 (Nrp1) | Nrp1, and VEGF | Reduced CNV was seen in Nrp1 knockout mice | Fernandez-Robredo et al., 2017 [116] |
| 6 | Platelet-activating factor (PFA) | PFA, PFA-receptor (PFA-R), macrophages, VEGF, MCP-1, and IL-6 | WEB2086, a novel PAF-R antagonist, inhibited CNV and experimentally induced subretinal fibrosis in mice | Zhang et al., 2013 [117] |
| 7 | Nucleoside reverse transcriptase inhibitors (NRTIs) | VEGF-A, and P2X7 receptor | Intravitreal injection of NRTIs, lamivudine, zidovudine, abacavir, and P2X7 antagonist A438079 reduced CNV in mice | Mizutani et al., 2015 [118] |
| 8 | RG7716 antibody | VEGF, and angiopoietin 2 | Phase II clinical trial underway. Phase I results indicated improvement in visual acuity in patients, and that RG7716 was safe | Chakravarthy et al., 2017 [26] |
| 9 | STAT3 signaling | Monocytes, macrophages, CX3CR1, HLA-DR, STAT3, VEGF, Suppressor of Cytokine Signalling 3 | Inhibition of STAT3 activation using LLL12-attenuated CNV in mice and intermediate monocytes (CD14+ CD16+) are activated in nAMD patients | Chen et al., 2016 [119] |
| 10 | TGF-β signaling | TGF-β, Smad2/3, VEGF, and TNF-α | Inhibition of TGF-β using a synthetic inhibitor, LY2157299 or Decorin, a natural TGF-β inhibitor significantly inhibited CNV in mice | Wang et al., 2017 [120] |
| 11 | Yes-associated protein (YAP) signaling | YAP, proliferating cell nuclear antigen (PCNA), CD31, VEGF | YAP small interfering RNA (siRNA) and ranibizumab treatment reduced VEGF and PCNA, reduced endothelial cell proliferation, and CNV formation in mice | Yan et al., 2018 [121] |
| 12 | Adeno-associated virus-mediated gene therapy with cartilage oligomeric matrix protein angiopoietin-1 (AAV2.COMP-Ang1) | VEGF, and hypoxia-inducible factor (HIF)-α | Subretinal injection of AAV2.COMP-Ang1 reduced VEGF levels and inhibited CNV in mice | Lambert et al., 2016 [122] |
| 13 | Fenofibric acid (Feno-FA) signaling | Feno-FA, VEGF, TNF-α, ICAM-1, and peroxisome proliferator–activated receptor-alpha (PPARα) | Feno-FA injections in mice suppressed neovascularization | Qiu et al., 2017 [123] |
| 14 | mTOR signaling | hypoxia-inducible gene RTP801, VEGF | A phase II clinical trial reported that the use of siRNA and PF-04523655 in combination with ranibizumab compared to ranibizumab alone improved vision in nAMD patients | Nguyen et al., 2012 [124] |
| 15 | Connective growth factor (CTGF) | CTGF, and ERK signaling | RXI-109, an inhibitor of CTGF, is designed to reduce retinal fibrosis in nAMD patients. Phase I clinical trial is currently underway | Kothary et al., 2010 [125]; ClinicalTrials.gov identifier: NCT02599064 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yerramothu, P. New Therapies of Neovascular AMD—Beyond Anti-VEGFs. Vision 2018, 2, 31. https://doi.org/10.3390/vision2030031
Yerramothu P. New Therapies of Neovascular AMD—Beyond Anti-VEGFs. Vision. 2018; 2(3):31. https://doi.org/10.3390/vision2030031
Chicago/Turabian StyleYerramothu, Praveen. 2018. "New Therapies of Neovascular AMD—Beyond Anti-VEGFs" Vision 2, no. 3: 31. https://doi.org/10.3390/vision2030031
APA StyleYerramothu, P. (2018). New Therapies of Neovascular AMD—Beyond Anti-VEGFs. Vision, 2(3), 31. https://doi.org/10.3390/vision2030031