
Theoretical Investigation of Glycine Micro-Solvated. Energy and NMR Spin Spin Coupling Constants Calculations


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
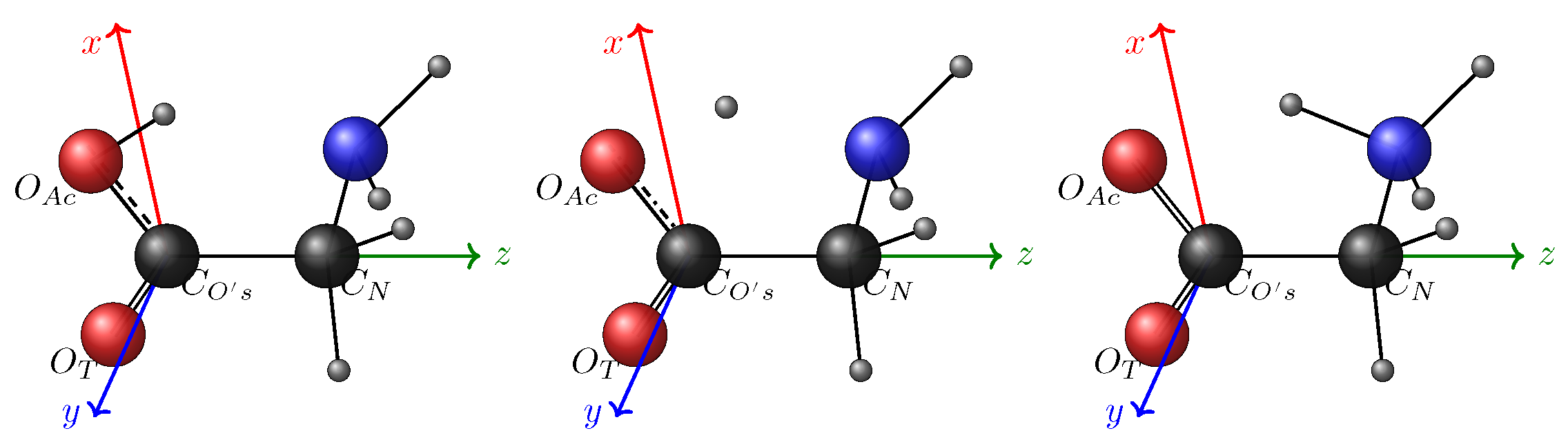
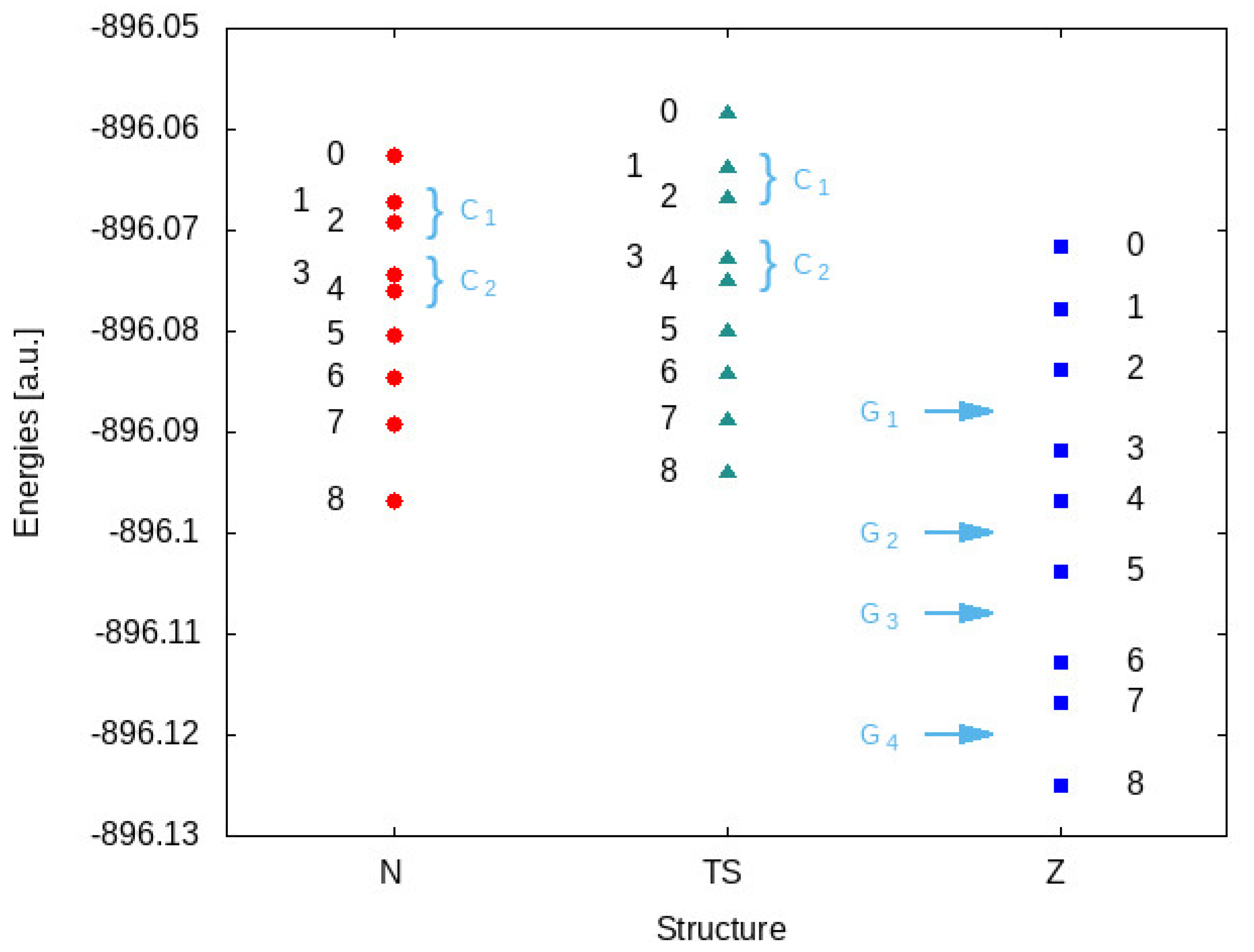
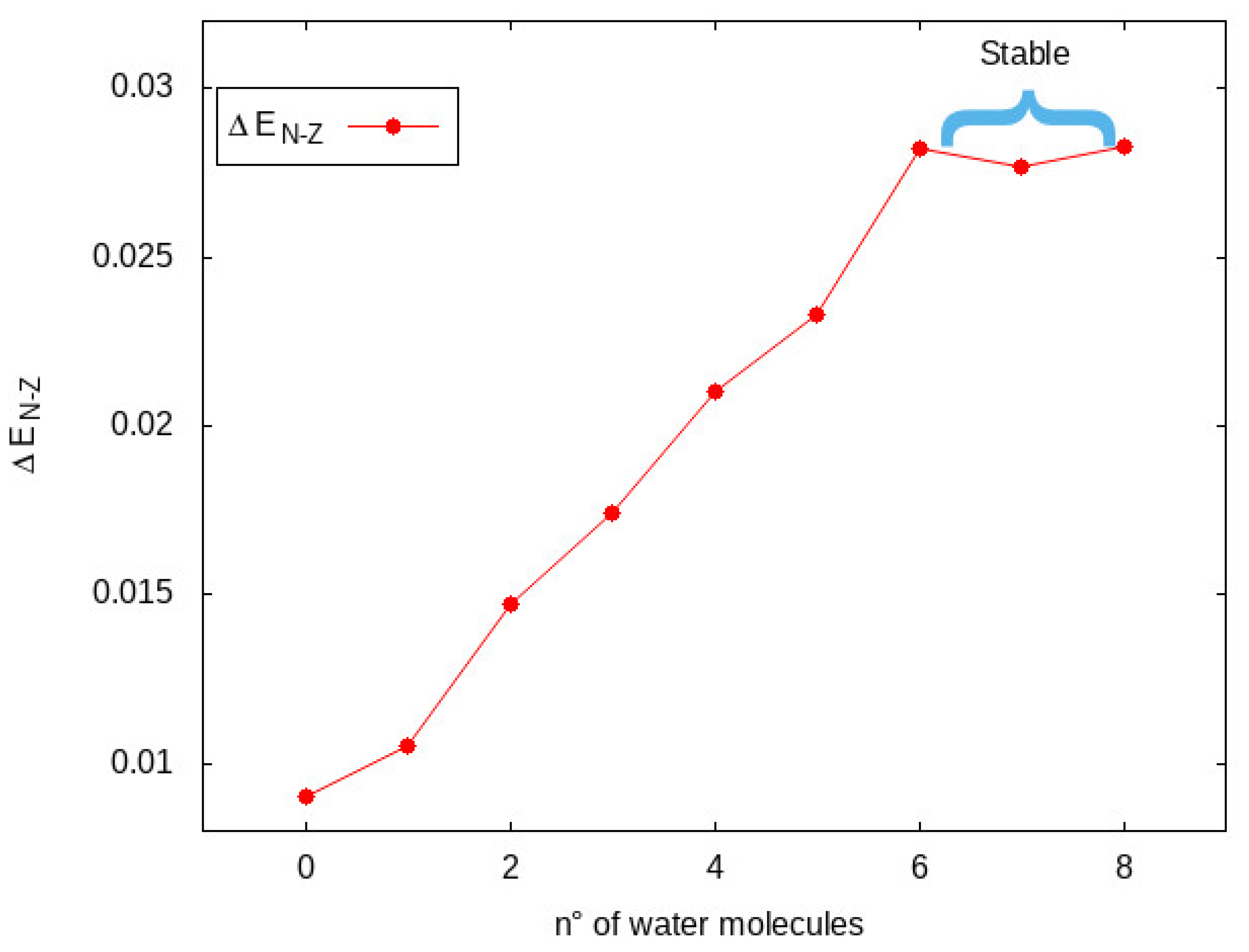
3.1. Basics Concepts
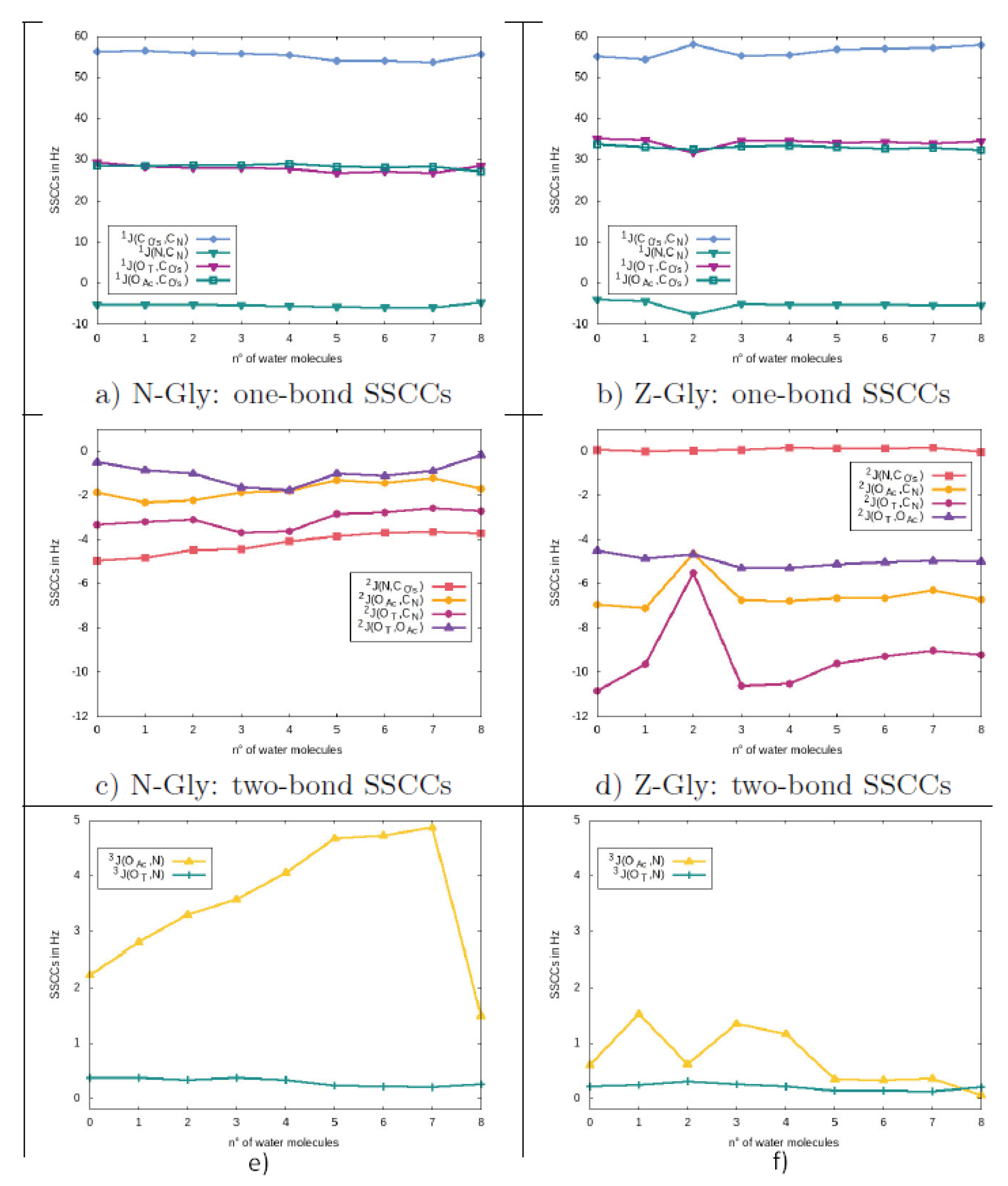
3.2. Spin–Spin Coupling Constants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jensen, J.H.; Gordon, M.S. On the Number of Water Molecules Necessary To Stabilize the Glycine Zwitterion. J. Am. Chem. Soc. 1995, 117, 8159–8170. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Ramos, A.; Smedarchina, Z.; Siebrand, W.; Zgierski, M.Z. A direct-dynamics study of the zwitterion-to-neutral interconversion of glycine in aqueous solution. J. Chem. Phys. 2000, 113, 9714–9721. [Google Scholar] [CrossRef]
- Karmacharya, R.; Antoniou, D.; Schwartz, S.D. Nonequilibrium Solvation and the Quantum Kramers Problem: Proton Transfer in Aqueous Glycine. J. Phys. Chem. A 2001, 105, 2563–2567. [Google Scholar] [CrossRef]
- Aikens, C.M.; Gordon, M.S. Incremental Solvation of Nonionized and Zwitterionic Glycine. J. Am. Chem. Soc. 2006, 128, 12835–12850. [Google Scholar] [CrossRef] [Green Version]
- Leung, K.; Rempe, S.B. Ab initio molecular dynamics study of glycine intramolecular proton transfer in water. J. Chem. Phys. 2005, 122, 184506. [Google Scholar] [CrossRef] [Green Version]
- Campo, M.G. Molecular dynamics simulation of glycine zwitterion in aqueous solution. J. Chem. Phys. 2006, 125, 114511. [Google Scholar] [CrossRef] [PubMed]
- Balabin, R.M. The First Step in Glycine Solvation: The Glycine-Water Complex. J. Phys. Chem. B 2010, 114, 15075–15078. [Google Scholar] [CrossRef]
- Takenaka, N.; Kitamura, Y.; Koyano, Y.; Asada, T.; Nagaoka, M. Reaction path optimization and vibrational frequency analysis via ab initio QM/MM free energy gradient (FEG) method: Application to isomerization process of glycine in aqueous solution. Theor. Chem. Acc. 2011, 130, 215–226. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ahn, D.S.; Park, S.W.; Lee, S. Gas phase hydration of amino acids and dipeptides: Effects on the relative stability of zwitterion vs. canonical conformers. RSC Adv. 2014, 4, 16352–16361. [Google Scholar] [CrossRef] [Green Version]
- Wada, G.; Tamura, E.; Okina, M.; Nacamura, M. On the ratio of zuitterion form to uncharged form of glycine at equilibrium in various aqueous media. Bull. Chem. Soc. Jpn. 1982, 55, 3064–3067. [Google Scholar] [CrossRef]
- Slifkin, M.A.; All, S.M. Thermodinamic parameters of the activation of glycine zwitterion protonation reactions. J. Mol. Liq. 1984, 28, 215–221. [Google Scholar] [CrossRef]
- Peteanu, L.A.; Levy, D.H. Spectroscopy of Complexes of Tryptamine and 3-Indolepropionic Acid with Various Solvents. J. Phys. Chem. 1988, 92, 6554–6561. [Google Scholar] [CrossRef]
- Xu, S.; Nilles, J.M.; Bowen, K.H. Zwitterion formation in hydrated amino acid, dipole bound anions: How many water molecules are required? J. Chem. Phys. 2003, 119, 10696. [Google Scholar] [CrossRef] [Green Version]
- Diken, E.G.; Hammer, N.I.; Johnson, M.A. Preparation and photoelectron spectrum of the glycine molecular anion: Assignment to a dipole-bound electron species with a high-dipole moment, non-zwitterionic form of the neutral core. J. Chem. Phys. 2004, 120, 9899–9902. [Google Scholar] [CrossRef]
- Nonose, S.; Iwaoka, S.; Mori, K.; Shibata, Y.; Fuke, K. Structures and reactions of hydrated biomolecular cluster ions. Eur. Phys. J. D 2005, 34, 315–319. [Google Scholar] [CrossRef]
- Alonso, J.L.; Cocinero, E.J.; Lesarri, A.; Sanz, M.E.; López, J.C. The Glycine-Water Complex. Angew. Chem. 2006, 118, 3551–3554. [Google Scholar] [CrossRef]
- da Silva, A.M.; Ghosh, A.; Chaudhuri, P. Effect of Hydrogen Bond Formation on the NMR Properties of Glycine-HCN Complexes. J. Phys. Chem. A 2013, 117, 10274–10285. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Canuto, S.P.P.F. NMR spin–spin coupling constants in hydrogen-bonded glycine clusters. Int. J. Quantum Chem. 2018. [Google Scholar] [CrossRef]
- Valverde, D.; da Costa Ludwig, Z.M.; da Costa, C.R.; Ludwig, V.; Georg, H.C. Zwitterionization of glycine in water environment: Stabilization mechanism and NMR spectral signatures. J. Chem. Phys. 2018, 148, 024305. [Google Scholar] [CrossRef]
- Arroyuelo, A.; Martin, O.A.; Scheraga, H.A.; Vila, J.A. Assessing the One-Bond Cα-H spin–spin Coupling Constants in Proteins: Pros and Cons of Different Approaches. J. Phys. Chem. B 2020, 124, 735–741. [Google Scholar] [CrossRef]
- Császár, A.G. Conformers of Gaseous Glycine. J. Am. Chem. Soc. 1992, 114, 9568–9575. [Google Scholar] [CrossRef]
- Godfrey, P.D.; Brown, R.D.; Rodgers, F.M. The missing conformers of glycine and alanine: Relaxation in seeded supersonic jets. J. Mol. Struct. 1996, 376, 65–81. [Google Scholar] [CrossRef]
- Sauer, S.P.A.; Oddershede, J.; Sabin, J.R. Directional Dependence of the Mean Excitation Energy and Spectral Moments of the Dipole Oscillator Strength Distribution of Glycine and Its Zwitterion. J. Phys. Chem. A 2006, 110, 8811–8817. [Google Scholar] [CrossRef]
- Caputo, M.C.; Provasi, P.F.; Sauer, S.P.A. The role of explicit solvent molecules in the calculation of NMR chemical shifts of glycine in water. Theor. Chem. Acc. 2018, 137, 88. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Krogb-Jespersen, K. The glycine zwitterion does not exist in the gas phase: Results from a detailed ab initio electronic structure study. Chem. Phys. Lett. 1992, 199, 261–266. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian- Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Kjær, H.; Sauer, S.P.A. Pople Style Basis Sets for the Calculation of NMR spin–spin Coupling Constants: The 6-31G-J and 6-311G-J Basis Sets. J. Chem. Theory Comput. 2011, 7, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibsom, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Lisong Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. Magn. Reson. Chem. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Miertus, S.; Scroco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Miertus, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B. New applications of integral equations methods for solvation continuum models: Ionic solutions and liquid crystals. J. Math. Chem. 1998, 23, 309–326. [Google Scholar] [CrossRef]
- Ramsey, N.F. Electron Coupled Interactions between Nuclear Spins in Molecules. Phys. Rev. 1953, 91, 303–307. [Google Scholar] [CrossRef]
- Autschbach, J.; Guennic, B.L. Analyzing and interpreting NMR spin–spin coupling constants using molecular orbitalcalculations. J. Chem. Educ. 2007, 84, 156–171. [Google Scholar] [CrossRef]
- Sauer, S.P.A. Molecular Electromagnetism: A Computational Chemistry Approach; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Helgaker, T.; Jaszuński, M.; Ruud, K. Ab Initio Methods for the Calculation of NMR Shielding and Indirect spin–spin Coupling Constants. Chem. Rev. 1999, 99, 293–352. [Google Scholar] [CrossRef]
- Krivdin, L.B.; Contreas, R.H. Recent advances in theoretical calculations of indirect spin–spin coupling constants. Annu. Rep. NMR Spectrosc. 2007, 61, 133–245. [Google Scholar]
- Vaara, J. Theory and computation of nuclear magnetic resonance parameters. Phys. Chem. Chem. Phys. 2007, 9, 5399–5418. [Google Scholar] [CrossRef] [PubMed]
- Helgaker, T.; Jaszunski, M.; Pecul, M. The quantum-chemical calculation of NMR indirect spin–spin coupling constants. Prog. Nucl. Magn. Reson. Spectrosc. 2008, 53, 249–268. [Google Scholar] [CrossRef]
- Helgaker, T.; Coriani, S.; Jørgensen, P.; Kristensen, K.; Olsen, J.; Ruud, K. Recent advances in wave function-based methods of molecular-property calculations. Chem. Rev. 2012, 112, 543–631. [Google Scholar]
- Sogn, J.A.; Craig, L.C.; Gibbons, W.A. Carbon-13-carbon-13 coupling constants in a series of carbon-13-enriched amino acids. J. Am. Chem. Soc. 1974, 96, 4694–4696. [Google Scholar] [CrossRef]
- Fermandjian, S.; Trandinh, S.; Savrda, J.; Sala, E.; Mermetbouvier, R.; Bricas, E.; Fromageot, P. 13C-nuclear magnetic resonance studies of 85% 13C-enriched amino acids and small peptides: pH effects on the chemical shifts, coupling constants, kinetics of cis-trans isomerisation and conformation aspects. Biochim. Biophys. Acta Gen. Subj. 1975, 399, 313–338. [Google Scholar] [CrossRef]
- Lichter, R.L.; Roberts, J.D. 15N Nuclear Magnetic Resonance Spectroscopy. XIII. Pyridine-15N1. J. Am. Chem. Soc. 1971, 93, 5218–5224. [Google Scholar] [CrossRef]
- Lemaitre, V.; Smith, M.E.; Watts, A. A review of oxygen-17 solid-state NMR of organic materials-towards biological applications. Solid State Nucl. Magn. Reson. 2004, 26, 215–235. [Google Scholar] [CrossRef]
- Sergeyev, N.M.; Sergeyeva, N.D.; Strelenko, Y.A.; Raynes, W.T. The 1H-2H, 17O-1H coupling constants and the 16O/18O induced proton isotope shift in water. Chem. Phys. Lett. 1997, 277, 142–146. [Google Scholar] [CrossRef]
- Halle, B.; Karlstrom, G. Prototropic charge migration in water. Part 1.-Rate constants in light and heavy water and in salt solution from oxygen-17 spin relaxation. J. Chem. Soc. Faraday Trans. 2 1983, 79, 1031–1046. [Google Scholar] [CrossRef]
- Broze, M.; Luz, Z. Oxygen-17 spin–spincoupling with manganese-55 and carbon-13. J. Phys. Chem. 1969, 73, 1600–1602. [Google Scholar] [CrossRef]
- Klemperer, W.G. 17O-NMR Spectroscopy as a Structural Probe. Angew. Chem. Int. Ed. Engl. 1978, 17, 246–254. [Google Scholar] [CrossRef]
- Amour, T.S.; Fiat, D. Oxygen-17 Magnetic Resonance. Bull. Magn. Reson. 1979, 1, 118–129. [Google Scholar]
- Gerothanassis, I.P. Oxygen-17 NMR spectroscopy: Basic principles and applications (Part I). Prog. Nucl. Magn. Reson. Spectrosc. 2010, 56, 95–197. [Google Scholar] [CrossRef]
- Gerothanassis, I.P. Oxygen-17 NMR spectroscopy: Basic principles and applications (part II). Prog. Nucl. Magn. Reson. Spectrosc. 2010, 57, 1–110. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H-Bond Sites | N/Z-Structure | Insertion Order |
|---|---|---|
| Ac | H-O-H ⋯ (OH)-C | 1, 6 |
| Am | HO ⋯ HN-C | 2, 4 |
| O | H-O-H ⋯ O=C | 3, 5, 7 |
| AmAc | C-O-H ⋯ (OH)-H ⋯ (NH)-C | 8 |
| Coupling | Molecule | 0 W | 8 W | PCM | ASEC-FEG | Exp. |
|---|---|---|---|---|---|---|
| N | 56.38 | 55.61 | ||||
| Z | 55.04 | 57.87 | 42.3 | 56.5 | 53.6 | |
| N | −5.24 | −4.82 | ||||
| Z | −4.09 | −5.40 | 0.5 | 3.3 | 6.2 | |
| N | 29.38 | 28.52 | ||||
| Z | 35.11 | 34.51 | 36.6 | 30.7 | ||
| N | 28.53 | 27.10 | ||||
| Z | 33.70 | 32.43 | 37.6 | 31.4 | ||
| N | −4.94 | −3.71 | ||||
| Z | 0.06 | −0.05 | ||||
| N | −1.86 | −1.69 | ||||
| Z | −6.94 | −6.70 | ||||
| N | −3.32 | −2.71 | ||||
| Z | −10.83 | −9.22 | ||||
| N | −0.51 | −0.17 | ||||
| Z | −4.50 | −4.99 | ||||
| N | 2.23 | 1.49 | ||||
| Z | 0.61 | 0.05 | ||||
| N | 0.37 | 0.25 | ||||
| Z | 0.22 | 0.20 |
| Coupling | Molecule | 4W–0W | 8W–4W | 8W–0W | |||
|---|---|---|---|---|---|---|---|
| FC | Total | FC | Total | FC | Total | ||
| N | −0.91 | −0.94 | 0.38 | 0.17 | −0.53 | −0.77 | |
| Z | 0.60 | 0.47 | 2.53 | 2.36 | 3.13 | 2.83 | |
| N | −0.45 | −0.35 | 0.77 | 0.77 | 0.31 | 0.42 | |
| Z | −1.17 | −1.23 | −0.15 | −0.09 | −1.32 | −1.31 | |
| N | −1.71 | −1.60 | 1.05 | 0.74 | −0.66 | −0.86 | |
| Z | −0.28 | −0.41 | −0.14 | −0.18 | −0.42 | −0.59 | |
| N | 0.35 | 0.43 | −1.81 | −1.87 | −1.46 | −1.44 | |
| Z | −0.32 | −0.23 | −1.35 | −1.02 | −1.67 | −1.26 | |
| N | 0.86 | 0.87 | 0.38 | 0.37 | 1.24 | 1.24 | |
| Z | 0.11 | 0.10 | −0.22 | −0.21 | −0.11 | −0.11 | |
| N | 0.03 | 0.06 | 0.12 | 0.11 | 0.15 | 0.17 | |
| Z | 0.09 | 0.18 | 0.03 | 0.07 | 0.12 | 0.25 | |
| N | −0.29 | −0.31 | 0.89 | 0.92 | 0.61 | 0.61 | |
| Z | 0.23 | 0.34 | 1.25 | 1.27 | 1.48 | 1.61 | |
| N | −1.36 | −1.25 | 1.44 | 1.58 | 0.08 | 0.33 | |
| Z | −0.83 | −0.78 | 0.44 | 0.30 | −0.40 | −0.48 | |
| N | 1.89 | 1.82 | −2.69 | −2.56 | −0.80 | −0.74 | |
| Z | 0.59 | 0.56 | −1.21 | −1.12 | −0.63 | −0.56 | |
| N | −0.06 | −0.04 | −0.05 | −0.08 | −0.10 | −0.12 | |
| Z | −0.03 | 0.00 | 0.03 | −0.02 | 0.00 | −0.02 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caputo, M.C.; Provasi, P.F. Theoretical Investigation of Glycine Micro-Solvated. Energy and NMR Spin Spin Coupling Constants Calculations. Sci 2021, 3, 41. https://doi.org/10.3390/sci3040041
Caputo MC, Provasi PF. Theoretical Investigation of Glycine Micro-Solvated. Energy and NMR Spin Spin Coupling Constants Calculations. Sci. 2021; 3(4):41. https://doi.org/10.3390/sci3040041
Chicago/Turabian StyleCaputo, Maria Cristina, and Patricio Federico Provasi. 2021. "Theoretical Investigation of Glycine Micro-Solvated. Energy and NMR Spin Spin Coupling Constants Calculations" Sci 3, no. 4: 41. https://doi.org/10.3390/sci3040041
APA StyleCaputo, M. C., & Provasi, P. F. (2021). Theoretical Investigation of Glycine Micro-Solvated. Energy and NMR Spin Spin Coupling Constants Calculations. Sci, 3(4), 41. https://doi.org/10.3390/sci3040041