Application of PCR-Based Tools to Explore Strongyloides Infection in People in Parts of Northern Australia

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
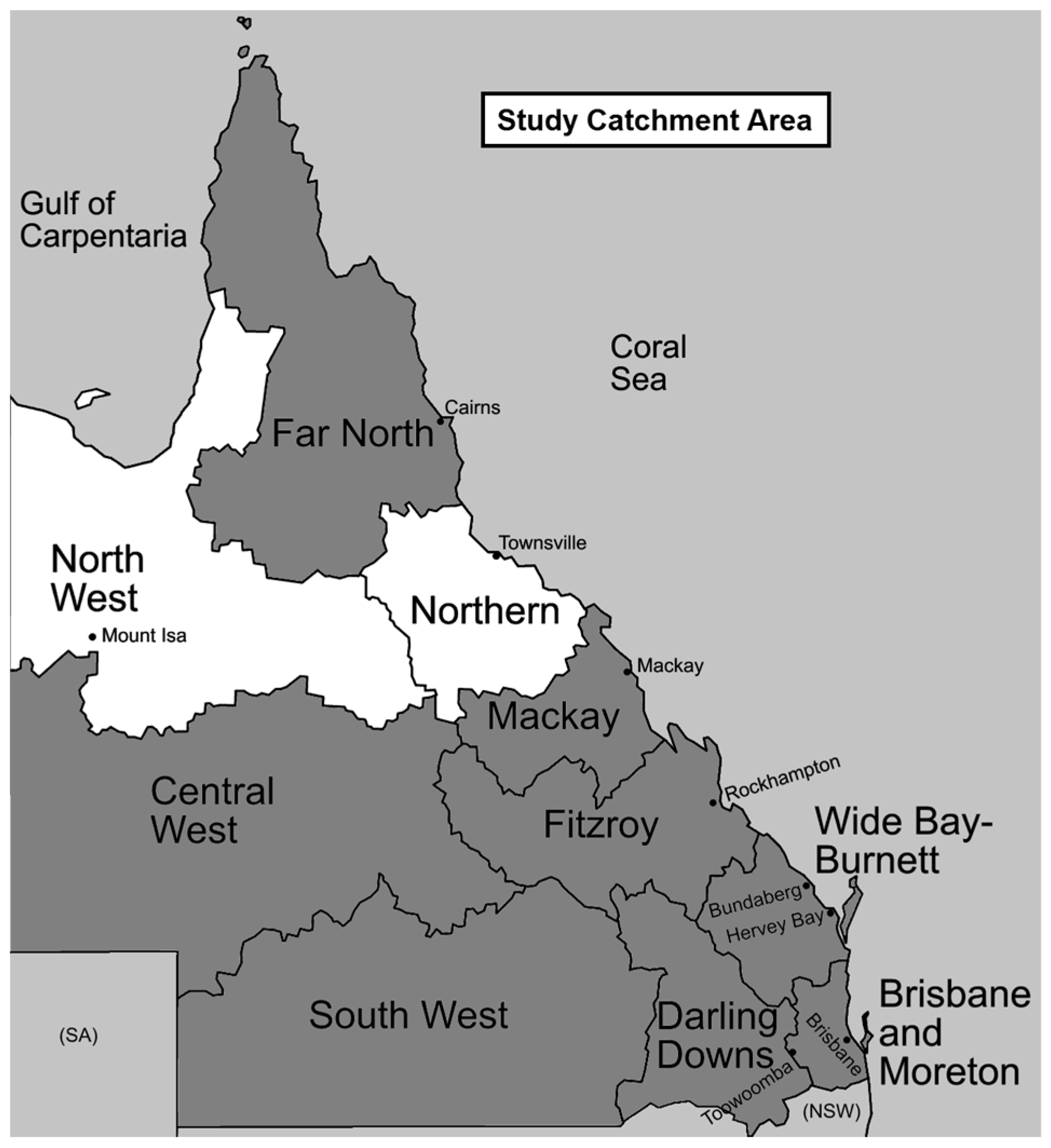
2.2. Specimen Collection and Study Site
2.3. Microscopy
2.4. DNA Extraction
2.5. Real-Time PCR
2.6. PCR-Based Single-Strand Conformation (SSCP) Analysis and Sequencing
2.7. Eosinophil Counts and Strongyloides Serology
2.8. Statistical Analysis
3. Results
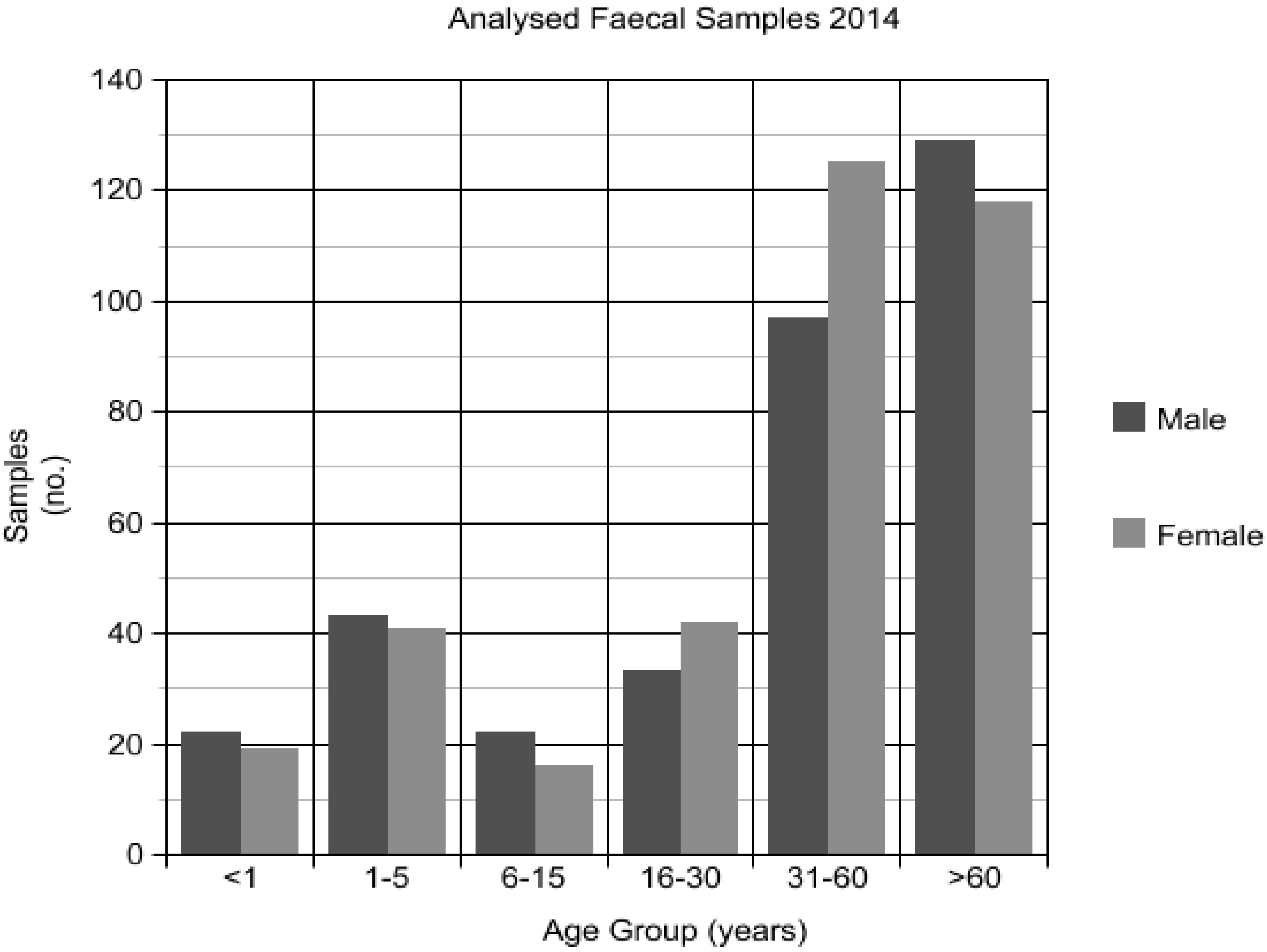
3.1. Patient Demographics and Specimen Selection
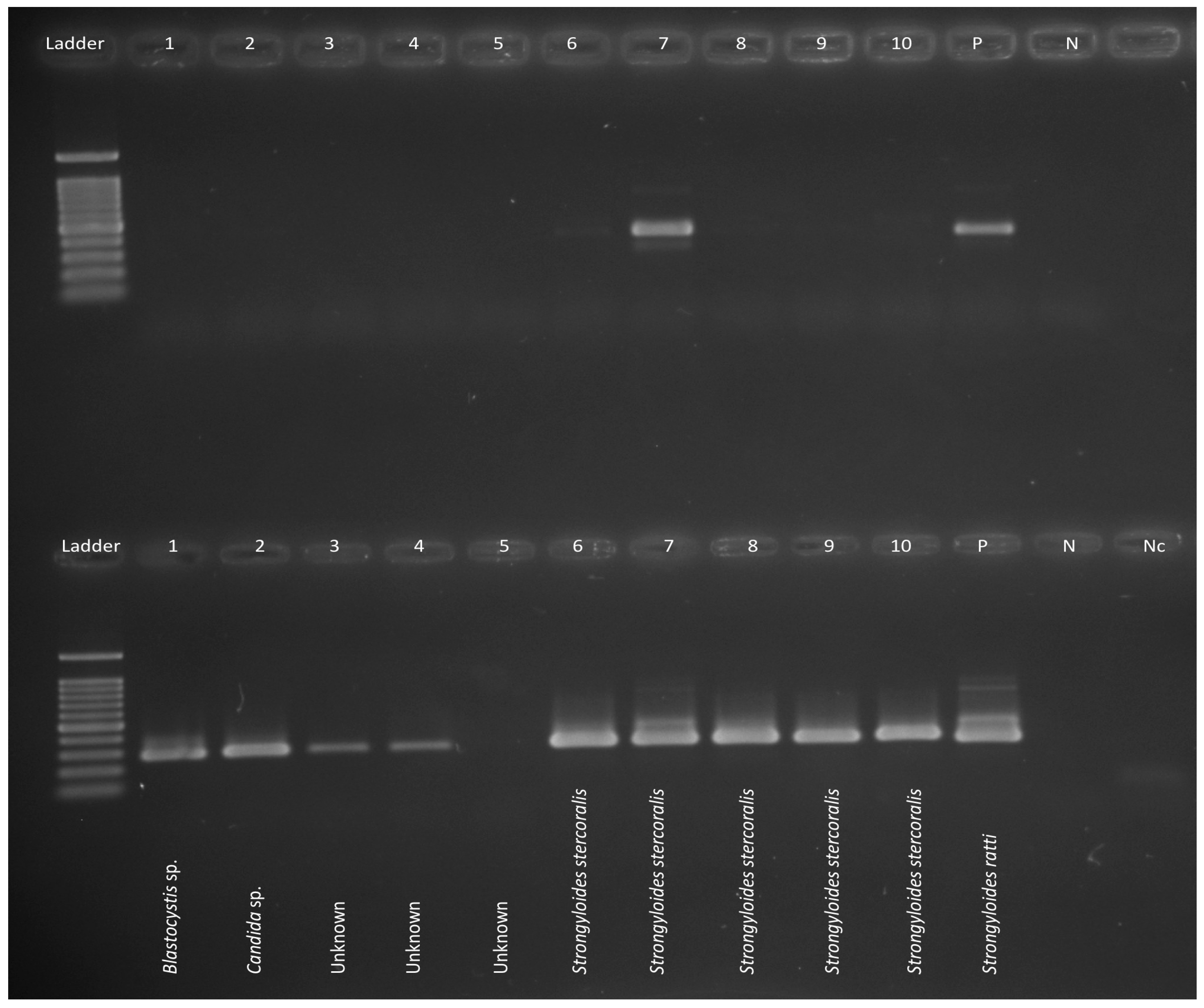
3.2. Microscopy, Harada-Mori Culture and Real-Time PCR Results
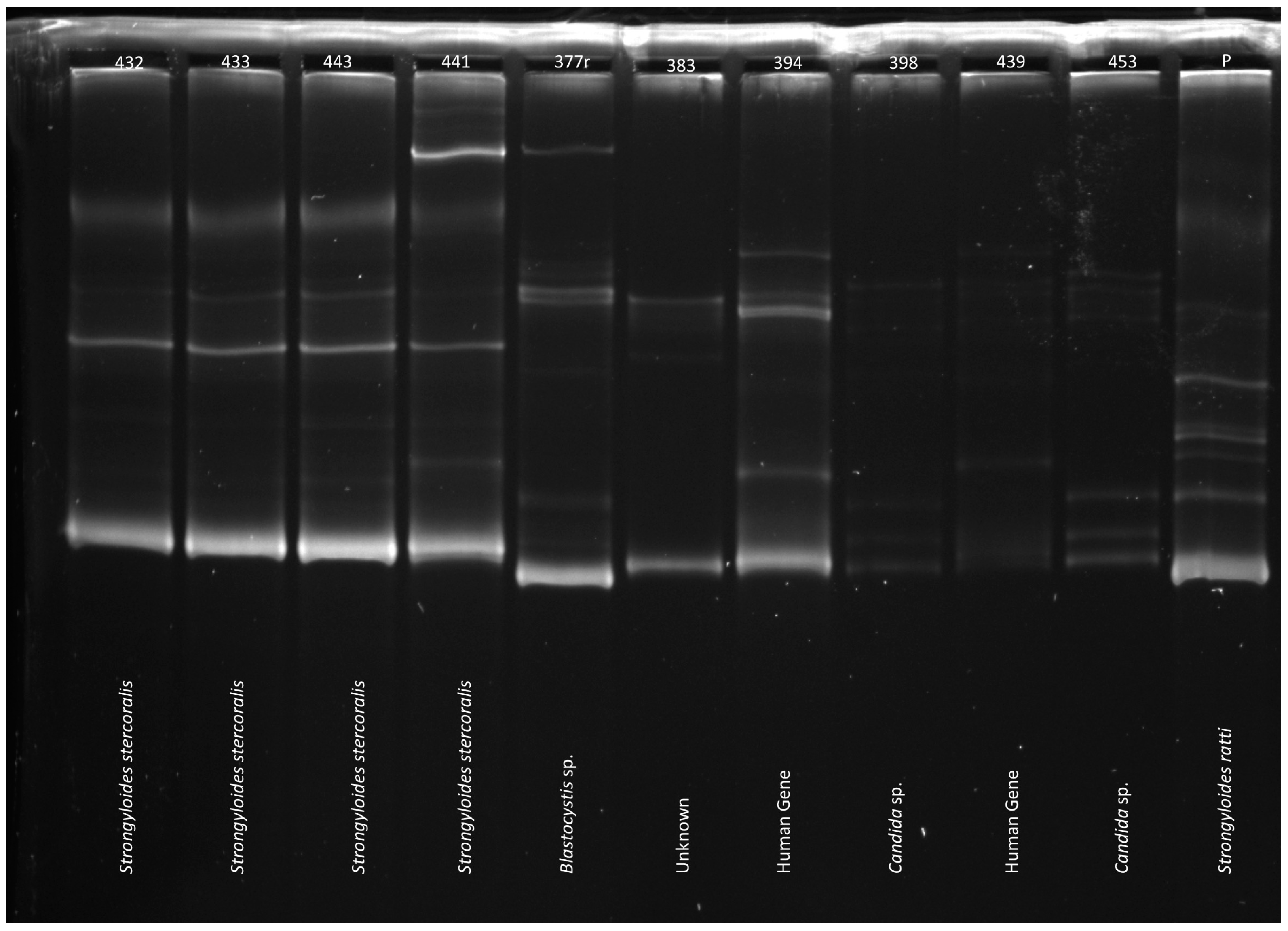
3.3. PCR-SSCP Analysis and Sequencing
3.4. Comparison of Serology and Eosinophilia with qPCR Results
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cook, G.C. Strongyloidiasis: A Major Roundworm Infection of Man; Grove, D.I., Ed.; Taylor & Francis: London, UK, 1989; ISBN 978-0-85066-732-5. [Google Scholar]
- Bisoffi, Z.; Buonfrate, D.; Montresor, A.; Requena-Méndez, A.; Muñoz, J.; Krolewiecki, A.J.; Gotuzzo, E.; Mena, M.A.; Chiodini, P.L.; Anselmi, M.; et al. Strongyloides stercoralis: A plea for action. PLoS Negl. Trop. Dis. 2013, 7, e2214. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.B.; Mojon, M. Improved diagnosis of Strongyloides stercoralis by seven consecutive stool specimens. Zent. Bakteriol. Mikrobiol. Hyg. A 1987, 263, 616–618. [Google Scholar] [CrossRef]
- Meloni, B.P.; Thompson, R.C.; Hopkins, R.M.; Reynoldson, J.A.; Gracey, M. The prevalence of Giardia and other intestinal parasites in children, dogs and cats from aboriginal communities in the Kimberley. Med. J. Aust. 1993, 158, 157–159. [Google Scholar] [PubMed]
- Reynoldson, J.A.; Behnke, J.M.; Pallant, L.J.; Macnish, M.G.; Gilbert, F.; Giles, S.; Spargo, R.J.; Thompson, R.C. Failure of pyrantel in treatment of human hookworm infections (Ancylostoma duodenale) in the Kimberley region of north west Australia. Acta Trop. 1997, 68, 301–312. [Google Scholar] [CrossRef]
- Fisher, D.; McCarry, F.; Currie, B. Strongyloidiasis in the Northern Territory. Under-recognised and under-treated? Med. J. Aust. 1993, 159, 88–90. [Google Scholar] [PubMed]
- Einsiedel, L.J.; Woodman, R.J. Two nations: Racial disparities in bloodstream infections recorded at Alice Springs Hospital, central Australia, 2001–2005. Med. J. Aust. 2010, 192, 567–571. [Google Scholar] [PubMed]
- Kukuruzovic, R.; Robins-Browne, R.M.; Anstey, N.M.; Brewster, D.R. Enteric pathogens, intestinal permeability and nitric oxide production in acute gastroenteritis. Pediatr. Infect. Dis. J. 2002, 21, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Johnston, F.H.; Morris, P.S.; Speare, R.; McCarthy, J.; Currie, B.; Ewald, D.; Page, W.; Dempsey, K. Strongyloidiasis: A review of the evidence for Australian practitioners. Aust. J. Rural Health 2005, 13, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Prociv, P.; Luke, R. Observations on strongyloidiasis in Queensland aboriginal communities. Med. J. Aust. 1993, 158, 160–163. [Google Scholar] [PubMed]
- Anamnart, W.; Pattanawongsa, A.; Intapan, P.M.; Maleewong, W. Albendazole stimulates the excretion of Strongyloides stercoralis larvae in stool specimens and enhances sensitivity for diagnosis of strongyloidiasis. J. Clin. Microbiol. 2010, 48, 4216–4220. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, M.S.; Harper, K.; Smith, N.; Verbanac, P.; Smith, J.W. Comparative evaluation of a modified zinc sulfate flotation technique. J. Clin. Microbiol. 1978, 7, 524–528. [Google Scholar] [PubMed]
- Steinmann, P.; Zhou, X.-N.; Du, Z.-W.; Jiang, J.-Y.; Wang, L.-B.; Wang, X.-Z.; Li, L.-H.; Marti, H.; Utzinger, J. Occurrence of Strongyloides stercoralis in Yunnan Province, China, and comparison of diagnostic methods. PLoS Negl. Trop. Dis. 2007, 1, e75. [Google Scholar] [CrossRef] [PubMed]
- Loukas, A. Neglected Tropical Diseases—Oceania; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-43148-2. [Google Scholar]
- Koga, K.; Kasuya, S.; Khamboonruang, C.; Sukavat, K.; Nakamura, Y.; Tani, S.; Ieda, M.; Tomita, K.; Tomita, S.; Hattan, N. An evaluation of the agar plate method for the detection of Strongyloides stercoralis in northern Thailand. J. Trop. Med. Hyg. 1990, 93, 183–188. [Google Scholar] [PubMed]
- Panosian, K.J.; Marone, P.; Edberg, S.C. Elucidation of Strongyloides stercoralis by bacterial-colony displacement. J. Clin. Microbiol. 1986, 24, 86–88. [Google Scholar] [PubMed]
- Arakaki, T.; Iwanaga, M.; Kinjo, F.; Saito, A.; Asato, R.; Ikeshiro, T. Efficacy of agar-plate culture in detection of Strongyloides stercoralis infection. J. Parasitol. 1990, 76, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Kasuya, S.; Ohtomo, H. How effective is the agar plate method for Strongyloides stercoralis? J. Parasitol. 1992, 78, 155–156. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, G.; Fernandes-Silva, E.; Alves, S.; Rocha, A.; Albuquerque, R.; Addiss, D. Patterns of detection of Strongyloides stercoralis in stool specimens: Implications for diagnosis and clinical trials. J. Clin. Microbiol. 1996, 34, 2569–2571. [Google Scholar] [PubMed]
- De J. Inês, E.; Souza, J.N.; Santos, R.C.; Souza, E.S.; Santos, F.L.; Silva, M.L.S.; Silva, M.P.; Teixeira, M.C.A.; Soares, N.M. Efficacy of parasitological methods for the diagnosis of Strongyloides stercoralis and hookworm in faecal specimens. Acta Trop. 2011, 120, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Herwaldt, B.L. Laboratory-acquired parasitic infections from accidental exposures. Clin. Microbiol. Rev. 2001, 14, 659–688. [Google Scholar] [CrossRef] [PubMed]
- De Kaminsky, R.G. Evaluation of three methods for laboratory diagnosis of Strongyloides stercoralis infection. J. Parasitol. 1993, 79, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Soulsby, H.M.; Hewagama, S.; Brady, S. Case series of four patients with strongyloides after occupational exposure. Med. J. Aust. 2012, 196, 444. [Google Scholar] [CrossRef] [PubMed]
- Cringoli, G. FLOTAC, a novel apparatus for a multivalent faecal egg count technique. Parassitologia 2006, 48, 381–384. [Google Scholar] [PubMed]
- Intapan, P.M.; Maleewong, W.; Wongsaroj, T.; Singthong, S.; Morakote, N. Comparison of the quantitative formalin ethyl acetate concentration technique and agar plate culture for diagnosis of human strongyloidiasis. J. Clin. Microbiol. 2005, 43, 1932–1933. [Google Scholar] [CrossRef] [PubMed]
- Glinz, D.; Silué, K.D.; Knopp, S.; Lohourignon, L.K.; Yao, K.P.; Steinmann, P.; Rinaldi, L.; Cringoli, G.; N’Goran, E.K.; Utzinger, J. Comparing diagnostic accuracy of Kato-Katz, Koga agar plate, ether-concentration, and FLOTAC for Schistosoma mansoni and soil-transmitted helminths. PLoS Negl. Trop. Dis. 2010, 4, e754. [Google Scholar] [CrossRef] [PubMed]
- Knopp, S.; Mgeni, A.F.; Khamis, I.S.; Steinmann, P.; Stothard, J.R.; Rollinson, D.; Marti, H.; Utzinger, J. Diagnosis of soil-transmitted helminths in the era of preventive chemotherapy: Effect of multiple stool sampling and use of different diagnostic techniques. PLoS Negl. Trop. Dis. 2008, 2, e331. [Google Scholar] [CrossRef] [PubMed]
- Marchi Blatt, J.; Cantos, G.A. Evaluation of techniques for the diagnosis of Strongyloides stercoralis in human immunodeficiency virus (HIV) positive and HIV negative individuals in the city of Itajaí, Brazil. Braz. J. Infect. Dis. 2003, 7, 402–408. [Google Scholar] [PubMed]
- Moody, A.; Rao, S.; Subirats, M. Evaluation of Parasep SF, Ether and Ethyl Acetate Free Faecal Concentrators; Apacaor: Berkshire, UK, 2013. [Google Scholar]
- Mounsey, K.; Kearns, T.; Rampton, M.; Llewellyn, S.; King, M.; Holt, D.; Currie, B.J.; Andrews, R.; Nutman, T.; McCarthy, J. Use of dried blood spots to define antibody response to the Strongyloides stercoralis recombinant antigen NIE. Acta Trop. 2014, 138, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Requena-Méndez, A.; Chiodini, P.; Bisoffi, Z.; Buonfrate, D.; Gotuzzo, E.; Muñoz, J. The laboratory diagnosis and follow up of strongyloidiasis: A systematic review. PLoS Negl. Trop. Dis. 2013, 7, e2002. [Google Scholar] [CrossRef] [PubMed]
- Page, W.A.; Dempsey, K.; McCarthy, J.S. Utility of serological follow-up of chronic strongyloidiasis after anthelminthic chemotherapy. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Schaffel, R.; Nucci, M.; Carvalho, E.; Braga, M.; Almeida, L.; Portugal, R.; Pulcheri, W. The value of an immunoenzymatic test (enzyme-linked immunosorbent assay) for the diagnosis of strongyloidiasis in patients immunosuppressed by hematologic malignancies. Am. J. Trop. Med. Hyg. 2001, 65, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Sudarshi, S.; Stümpfle, R.; Armstrong, M.; Ellman, T.; Parton, S.; Krishnan, P.; Chiodini, P.L.; Whitty, C.J.M. Clinical presentation and diagnostic sensitivity of laboratory tests for Strongyloides stercoralis in travellers compared with immigrants in a non-endemic country. Trop. Med. Int. Health 2003, 8, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Verweij, J.J.; Canales, M.; Polman, K.; Ziem, J.; Brienen, E.A.T.; Polderman, A.M.; van Lieshout, L. Molecular diagnosis of Strongyloides stercoralis in faecal samples using real-time PCR. Trans. R. Soc. Trop. Med. Hyg. 2009, 103, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.R.; James, G.; Sultana, Y.; Ginn, A.N.; Outhred, A.C.; Kong, F.; Verweij, J.J.; Iredell, J.R.; Chen, S.C.-A.; Lee, R. A loop-mediated isothermal amplification (LAMP) assay for Strongyloides stercoralis in stool that uses a visual detection method with SYTO-82 fluorescent dye. Am. J. Trop. Med. Hyg. 2014, 90, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Gasser, R.B.; Hu, M.; Chilton, N.B.; Campbell, B.E.; Jex, A.J.; Otranto, D.; Cafarchia, C.; Beveridge, I.; Zhu, X. Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat. Protoc. 2006, 1, 3121–3128. [Google Scholar] [CrossRef] [PubMed]
- Queensland Government. Queensland Regional Profiles. Queensland Government Statistician’s Office. 2016. Available online: http://statistics.qgso.qld.gov.au/qld-regional-profiles (accessed on 4 January 2016).
- World Health Organization. CCTA/WHO African Conference on Ancylostomiasis; World Health Organization Technical Report Series No. 255; World Health Organization: Geneva, Switzerland, 1963. [Google Scholar]
- Sultana, Y.; Jeoffreys, N.; Watts, M.R.; Gilbert, G.L.; Lee, R. Real-time polymerase chain reaction for detection of Strongyloides stercoralis in stool. Am. J. Trop. Med. Hyg. 2013, 88, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Ten Hove, R.J.; van Esbroeck, M.; Vervoort, T.; van den Ende, J.; van Lieshout, L.; Verweij, J.J. Molecular diagnostics of intestinal parasites in returning travellers. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Sithithaworn, P.; Srisawangwong, T.; Tesana, S.; Daenseekaew, W.; Sithithaworn, J.; Fujimaki, Y.; Ando, K. Epidemiology of Strongyloides stercoralis in north-east Thailand: Application of the agar plate culture technique compared with the enzyme-linked immunosorbent assay. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 398–402. [Google Scholar] [CrossRef]
- Naidu, P.; Yanow, S.K.; Kowalewska-Grochowska, K.T. Eosinophilia: A poor predictor of Strongyloides infection in refugees. Can. J. Infect. Dis. Med. Microbiol. 2013, 24, 93–96. [Google Scholar] [PubMed]
- Schär, F.; Odermatt, P.; Khieu, V.; Panning, M.; Duong, S.; Muth, S.; Marti, H.; Kramme, S. Evaluation of real-time PCR for Strongyloides stercoralis and hookworm as diagnostic tool in asymptomatic schoolchildren in Cambodia. Acta Trop. 2013, 126, 89–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitta, R.B.; Malta, F.M.; Pinho, J.R.; Chieffi, P.P.; Gryschek, R.C.B.; Paula, F.M. Conventional PCR for molecular diagnosis of human strongyloidiasis. Parasitology 2014, 141, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Sharifdini, M.; Mirhendi, H.; Ashrafi, K.; Hosseini, M.; Mohebali, M.; Khodadadi, H.; Kia, E.B. Comparison of nested polymerase chain reaction and real-time polymerase chain reaction with parasitological methods for detection of Strongyloides stercoralis in human fecal samples. Am. J. Trop. Med. Hyg. 2015, 93, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Saugar, J.M.; Merino, F.J.; Martín-Rabadán, P.; Fernández-Soto, P.; Ortega, S.; Gárate, T.; Rodríguez, E. Application of real-time PCR for the detection of Strongyloides spp. in clinical samples in a reference center in Spain. Acta Trop. 2015, 142, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, S.; Inpankaew, T.; Nery, S.V.; Gray, D.J.; Verweij, J.J.; Clements, A.C.A.; Gomes, S.J.; Traub, R.; McCarthy, J.S. Application of a multiplex quantitative PCR to assess prevalence and intensity of intestinal parasite infections in a controlled clinical trial. PLoS Negl. Trop. Dis. 2016, 10, e0004380. [Google Scholar] [CrossRef] [PubMed]
- Polz, M.F.; Cavanaugh, C.M. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [Google Scholar] [PubMed]
- Hunt, V.L.; Tsai, I.J.; Coghlan, A.; Reid, A.J.; Holroyd, N.; Foth, B.J.; Tracey, A.; Cotton, J.A.; Stanley, E.J.; Beasley, H.; et al. The genomic basis of parasitism in the Strongyloides clade of nematodes. Nat. Genet. 2016, 48, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Janwan, P.; Intapan, P.M.; Thanchomnang, T.; Lulitanond, V.; Anamnart, W.; Maleewong, W. Rapid detection of Opisthorchis viverrini and Strongyloides stercoralis in human fecal samples using a duplex real-time PCR and melting curve analysis. Parasitol. Res. 2011, 109, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Caraguel, C.G.B.; Stryhn, H.; Gagné, N.; Dohoo, I.R.; Hammell, K.L. Selection of a cutoff value for real-time polymerase chain reaction results to fit a diagnostic purpose: Analytical and epidemiologic approaches. J. Vet. Diagn. Investig. 2011, 23, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Sato, H.; Fujita, S.; Nguema, P.P.M.; Nobusue, K.; Miyagi, K.; Kooriyama, T.; Takenoshita, Y.; Noda, S.; Sato, A.; et al. Molecular identification of the causative agent of human strongyloidiasis acquired in Tanzania: Dispersal and diversity of Strongyloides spp. and their hosts. Parasitol. Int. 2010, 59, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Shiwaku, K.; Chigusa, Y.; Kadosaka, T.; Kaneko, K. Factors influencing development of free-living generations of Strongyloides stercoralis. Parasitology 1988, 97 Pt 1, 129–138. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (Years) | Gender | Place of Residence | Region | Real-time PCR | Mean Ct Value | Nested PCR | Sequence Identity/SSCP Homology | Strongyloides Serology | Eosinophil Count (×109/L) | Parasites Detected by FEAC Microscopy |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | F | Doomadgee * | NW | Positive | 18.77 | Detected | S. stercoralis | nd | nd | S. stercoralis |
| 2 | F | Doomadgee * | NW | Positive | 19.16 | Detected | S. stercoralis | nd | 0.57 | S. stercoralis |
| 51 | F | Doomadgee | NW | Positive | 23.72 | Detected | S. stercoralis | Reactive (3.1) | 0.43 | S. stercoralis |
| 41 | F | Doomadgee | NW | Positive | 25.66 | Detected | S. stercoralis | Reactive (1.6) | 0.93 | |
| 2 | F | Doomadgee * | NW | Positive | 31.13 | Detected | S. stercoralis | nd | 3.69 | S. stercoralis |
| 9 | M | Doomadgee | NW | Positive | 35.25 | Detected | H. nana | nd | 2.84 | S. stercoralis, H. nana, B. hominis, E. nana |
| 70 | M | Aurukun * | NW | Positive | 27.44 | Detected | S. stercoralis | nd | 0.89 | B. hominis |
| 70 | M | Aurukun * | NW | Positive | 28.00 | Detected | S. stercoralis | nd | 0.72 | B. hominis |
| 28 | M | Mount Isa | NW | Positive | 28.10 | Detected | S. stercoralis | nd | nd | |
| 66 | M | Mount Isa | NW | Positive | 36.27 | Detected | B. hominis | nd | 0.02 | B. hominis |
| 30 | M | Mount Isa | NW | Positive | 36.41 | Detected | S. stercoralis | nd | 0.41 | |
| 61 | M | Magnetic Island | NTH | Positive | 31.15 | Detected | S. stercoralis | nd | nd | |
| 28 | M | Magnetic Island | NTH | Positive | 35.69 | Detected | S. stercoralis | nd | 0.30 | |
| 64 | F | Magnetic Island | NTH | Positive | 35.93 | Detected | Human gene | nd | 0.60 | |
| 51 | F | Magnetic Island | NTH | Positive | 36.12 | Detected | S. stercoralis | nd | nd | B. hominis |
| 36 | M | Townsville | NTH | Positive | 31.86 | Detected | S. stercoralis | Reactive (4.0) | 1.27 | B. hominis |
| 70 | M | Townsville | NTH | Positive | 34.12 | Detected | S. stercoralis | nd | 0.13 | |
| 40 | M | Townsville | NTH | Positive | 36.10 | Detected | S. stercoralis | Reactive (3.1) | 1.48 | H. nana, B. hominis, Entamoeba coli |
| <1 | F | Townsville | NTH | Positive | 36.13 | Detected | S. stercoralis | nd | 0.00 | |
| 58 | M | Townsville | NTH | Positive | 36.57 | Detected | S. stercoralis | nd | 0.10 | |
| <1 | M | Townsville | NTH | Positive | 37.08 | Detected | S. stercoralis | nd | nd | |
| 14 | F | Townsville | NTH | Positive | 37.31 | Detected | S. stercoralis | nd | nd | |
| 39 | F | Townsville | NTH | Positive | 38.51 | Not Detected | nd | 0.08 | ||
| 69 | F | Townsville | NTH | Positive | 38.97 | Detected | Candida sp. | nd | 0.46 | |
| 51 | F | Townsville | NTH | Positive | 39.33 | Detected | Candida sp. | nd | 0.20 | |
| 43 | F | Jundah | CW | Positive | 34.32 | Not Detected | nd | 0.04 | B. hominis | |
| 67 | F | Mornington Island | NW | Positive | 35.08 | Detected | B. hominis | Reactive (4.1) | 2.14 | |
| 1 | M | Mornington Island | NW | Positive | 36.96 | Detected | Human gene | nd | 8.04 | G. intestinalis |
| 35 | M | Normanton | NW | Positive | 35.81 | Detected | S. stercoralis | nd | 1.90 | |
| 36 | M | Palm Island | NTH | Positive | 36.53 | Detected | S. stercoralis | nd | 0.25 | |
| <1 y | M | Palm Island | NTH | Positive | 38.37 | Not Detected | nd | nd | ||
| 1 | M | Palm Island ^ | NTH | Positive | 38.48 | Detected | Human gene | nd | nd | |
| 20 | F | Palm Island | NTH | Positive | 38.84 | Detected | S. stercoralis | nd | 0.62 | |
| 78 | M | Ayr | NTH | Positive | 36.63 | Detected | S. stercoralis | nd | 0.14 | |
| 1 | M | Cloncurry | NW | Positive | 36.81 | Not Detected | nd | nd | B. hominis, E. nana | |
| 94 | F | Home Hill | NTH | Positive | 36.95 | Not Detected | nd | 0.12 | B. hominis | |
| 80 | F | Greenvale | NTH | Positive | 37.01 | Detected | S. stercoralis | nd | 0.09 | |
| 11 | F | Charters Towers | NTH | Positive | 37.15 | Detected | S. stercoralis | nd | nd | |
| 79 | M | Hughenden | NW | Positive | 37.66 | Detected | Saccharomyces sp. | nd | 1.05 | |
| 27 | M | Camooweal | NW | Positive | 38.12 | Detected | Saccharomyces sp. | nd | 0.25 | G. intestinalis |
| 84 | M | Ingham | NTH | Positive | 38.40 | Not Detected | nd | 0.09 | B. hominis | |
| 63 | F | Ingham | NTH | Positive | 38.53 | Not Detected | nd | 0.09 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robertson, G.J.; Koehler, A.V.; Gasser, R.B.; Watts, M.; Norton, R.; Bradbury, R.S. Application of PCR-Based Tools to Explore Strongyloides Infection in People in Parts of Northern Australia. Trop. Med. Infect. Dis. 2017, 2, 62. https://doi.org/10.3390/tropicalmed2040062
Robertson GJ, Koehler AV, Gasser RB, Watts M, Norton R, Bradbury RS. Application of PCR-Based Tools to Explore Strongyloides Infection in People in Parts of Northern Australia. Tropical Medicine and Infectious Disease. 2017; 2(4):62. https://doi.org/10.3390/tropicalmed2040062
Chicago/Turabian StyleRobertson, Gemma J., Anson V. Koehler, Robin B. Gasser, Matthew Watts, Robert Norton, and Richard S. Bradbury. 2017. "Application of PCR-Based Tools to Explore Strongyloides Infection in People in Parts of Northern Australia" Tropical Medicine and Infectious Disease 2, no. 4: 62. https://doi.org/10.3390/tropicalmed2040062
APA StyleRobertson, G. J., Koehler, A. V., Gasser, R. B., Watts, M., Norton, R., & Bradbury, R. S. (2017). Application of PCR-Based Tools to Explore Strongyloides Infection in People in Parts of Northern Australia. Tropical Medicine and Infectious Disease, 2(4), 62. https://doi.org/10.3390/tropicalmed2040062