The Emergence of Chikungunya ECSA Lineage in a Mayaro Endemic Region on the Southern Border of the Amazon Forest

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site
2.2. Sample Collection
2.3. Viral Detection—RT-PCR Assays
2.4. Viral Detection—Isolation
2.5. Viral Genome Sequencing
2.6. Phylogenetic Analysis
2.7. Ethical Approval
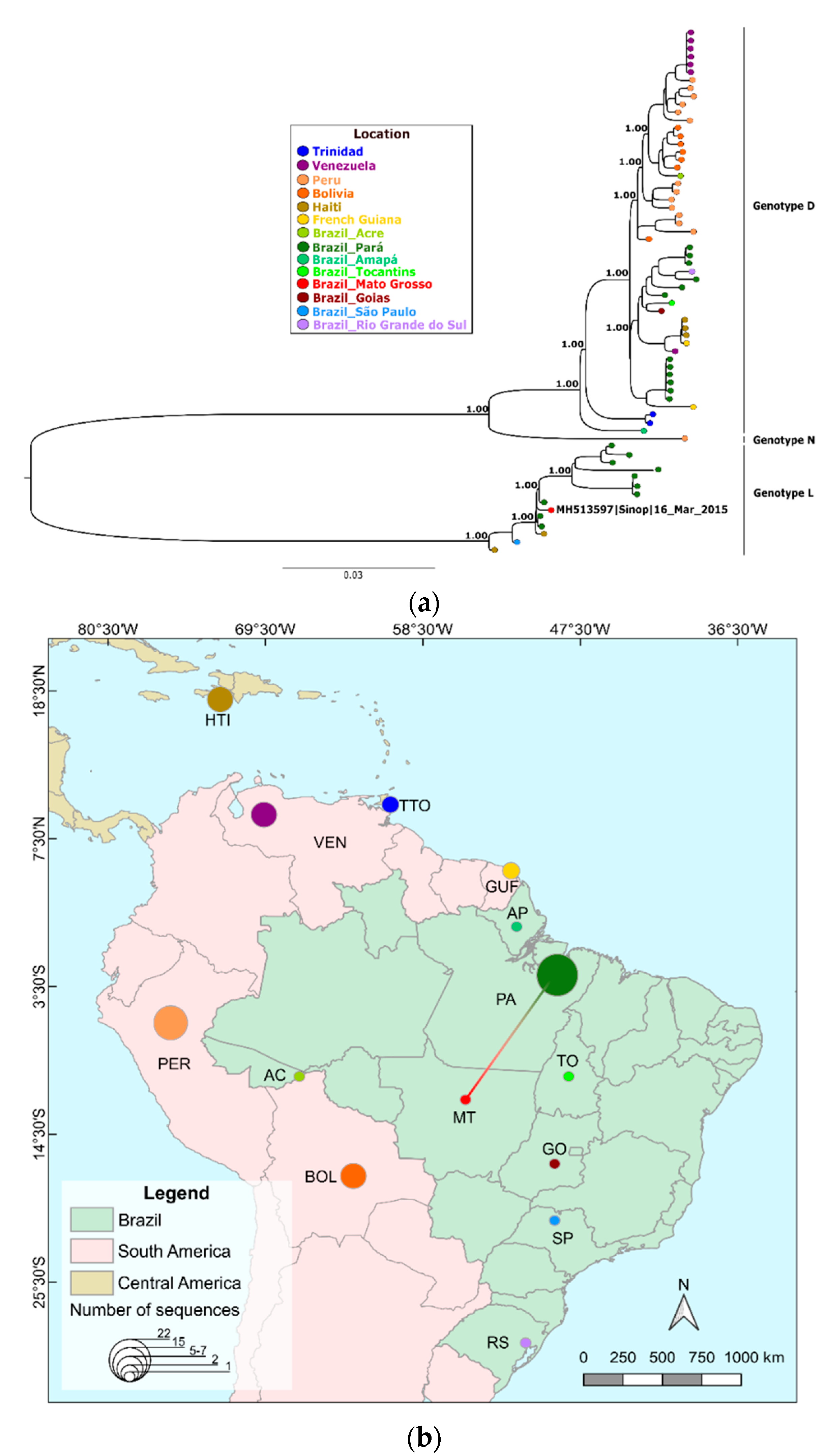
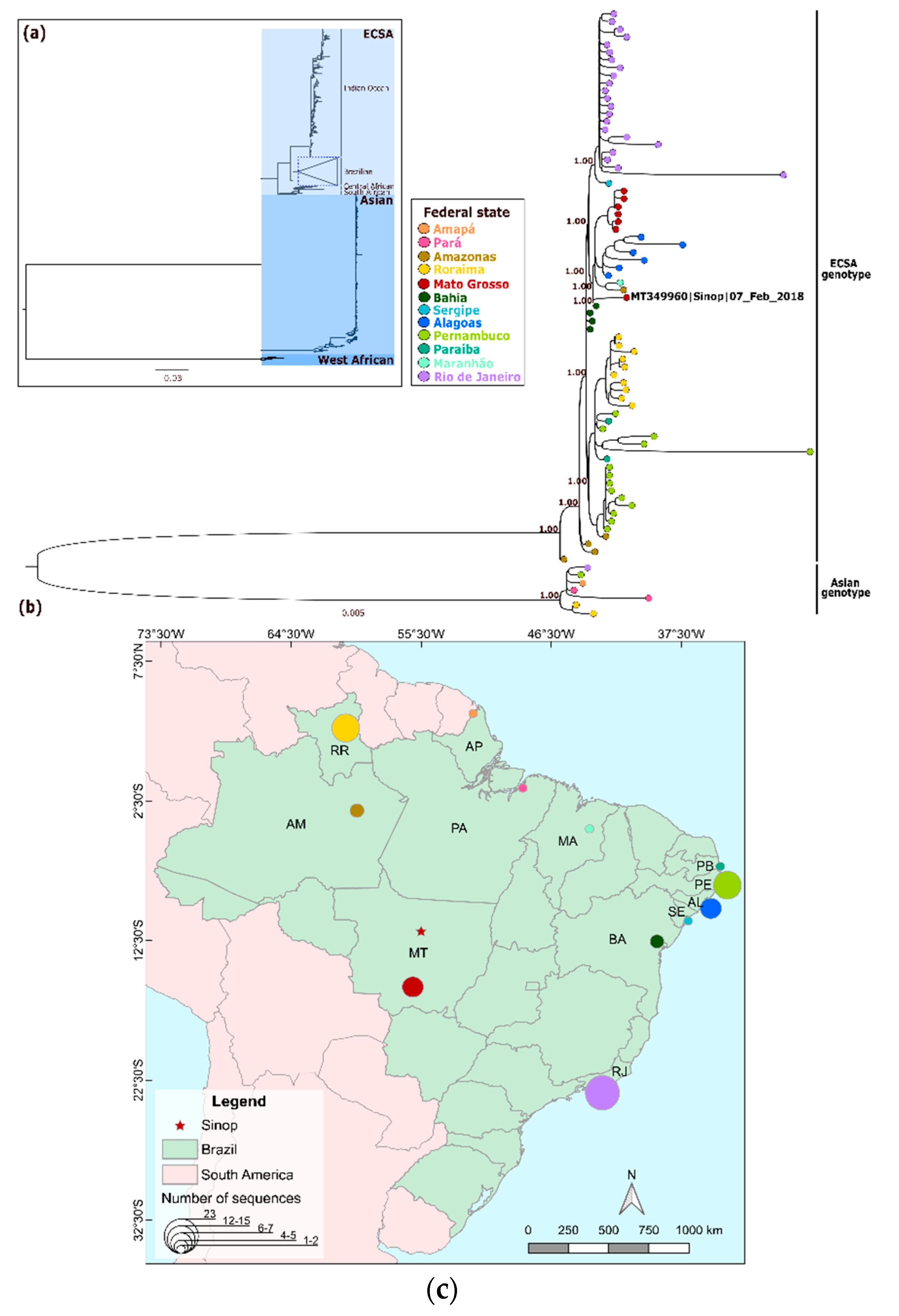
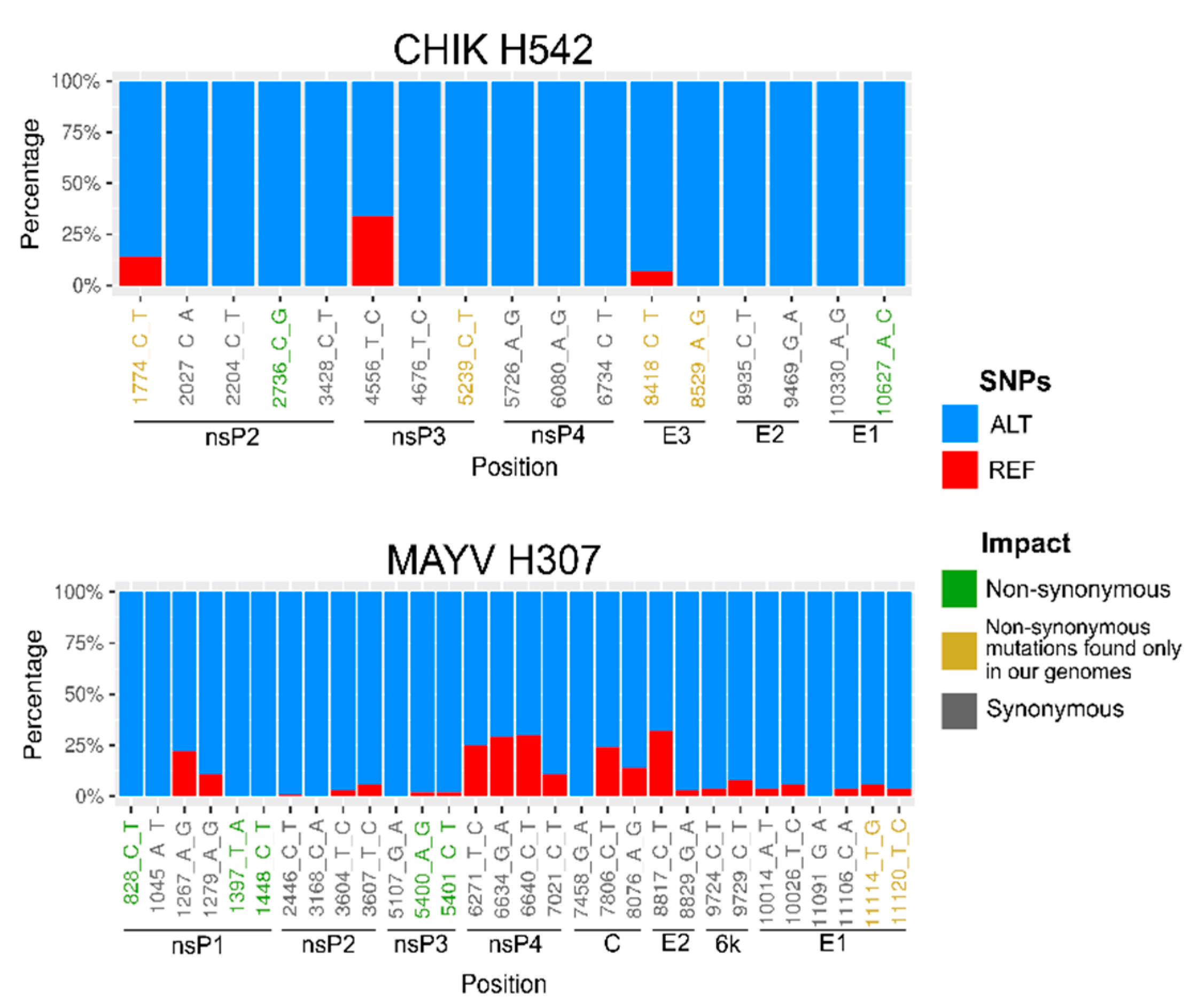
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ebel, G.D. Promiscuous viruses—how do viruses survive multiple unrelated hosts? Curr. Opin. Virol. 2017, 23, 125–129. [Google Scholar] [CrossRef]
- Marcondes, C.B.; Contigiani, M.; Gleiser, R.M. Emergent and Reemergent Arboviruses in South America and the Caribbean: Why So Many and Why Now? J. Med. Entomol. 2017, 54, 509–532. [Google Scholar] [CrossRef]
- World Health Organization Dengue and severe dengue. Available online: https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (accessed on 30 April 2020).
- Rocco, I.M.; Santos, C.L.S.; Bisordi, I.; Petrella, S.M.C.N.; Pereira, L.E.; Souza, R.P.; Coimbra, T.L.M.; Bessa, T.A.F.; Oshiro, F.M.; Lima, L.B.Q.; et al. St. Louis encephalitis virus: First isolation from a human in São Paulo State, Brazil. Rev. Inst. Med. Trop. Sao Paulo 2005, 47, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Mondini, A.; Bronzoni, R.V.d.M.; Cardeal, I.L.S.; dos Santos, T.M.I.L.; Lázaro, E.; Nunes, S.H.P.; Silva, G.C.D.; Madrid, M.C.F.S.; Rahal, P.; Figueiredo, L.T.; et al. Simultaneous infection by DENV-3 and SLEV in Brazil. J. Clin. Virol. 2007, 40, 84–86. [Google Scholar] [CrossRef]
- Vieira, M.A.C.e.S.; Aguiar, A.d.A.X.; De Borba, A.S.; Guimarães, H.C.L.; Eulálio, K.D.; de Albuquerque-Neto, L.L.; Salmito, M.d.A.; Lima, O.B. West Nile fever in Brazil: Sporadic case, silent endemic disease or epidemic in its initial stages? Rev. Inst. Med. Trop. Sao Paulo 2015, 57, 276. [Google Scholar] [CrossRef] [Green Version]
- Martins, L.C.; Da Silva, E.V.P.; Casseb, L.M.N.; Da Silva, S.P.; Cruz, A.C.R.; De Sousa Pantoja, J.A.; De Almeida Medeiros, D.B.; Filho, A.J.M.; Da Cruz, E.D.R.M.; De Araújo, M.T.F.; et al. First isolation of west nile virus in brazil. Mem. Inst. Oswaldo Cruz 2019, 114. [Google Scholar] [CrossRef] [PubMed]
- Terzian, A.C.B.; Auguste, A.J.; Vedovello, D.; Ferreira, M.U.; Da Silva-Nunes, M.; Sperança, M.A.; Suzuki, R.B.; Juncansen, C.; Jo, J.P.A.; Weaver, S.C.; et al. Isolation and characterization of Mayaro virus from a human in Acre, Brazil. Am. J. Trop. Med. Hyg. 2015, 92, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Vieira, C.J.d.S.P.; da Silva, D.J.F.; Barreto, E.S.; Siqueira, C.E.H.; Colombo, T.E.; Ozanic, K.; Schmidt, D.J.; Drumond, B.P.; Mondini, A.; Nogueira, M.L.; et al. Detection of Mayaro virus infections during a dengue outbreak in Mato Grosso, Brazil. Acta Trop. 2015, 147, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Dietrich, I.; Desiree LaBeaud, A.; Lindsay, S.W.; Musa, A.; Weaver, S.C. Risks and challenges of arboviral diseases in Sudan: The urgent need for actions. Viruses 2020, 12, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, M.; Dobler, G. Emergence of zoonotic arboviruses by animal trade and migration. Parasites and Vectors 2010, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.Y.; Balasuriya, U.B.R.; Lee, C. Zoonotic encephalitides caused by arboviruses: Transmission and epidemiology of alphaviruses and flaviviruses. Clin. Exp. Vaccine Res. 2014, 3, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, L.I.; Vignuzzi, M. Arthritogenic alphaviruses: A worldwide emerging threat? Microorganisms 2019, 7, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casals, J.; Whitman, L. Mayaro virus: A new human disease agent. I. Relationship to other arbor viruses. Am. J. Trop. Med. Hyg. 1957, 6, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.J.; Haddow, A.J. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952–1953. Trans. R. Soc. Trop. Med. Hyg. 1957, 51, 238–240. [Google Scholar] [CrossRef]
- Lorenz, C.; Freitas Ribeiro, A.; Chiaravalloti-Neto, F. Mayaro virus distribution in South America. Acta Trop. 2019, 198. [Google Scholar] [CrossRef]
- Ganjian, N.; Riviere-Cinnamond, A. Mayaro virus in Latin America and the Caribbean. Rev. Panam. Salud Pública 2020, 44, 1. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Puri, V.; Fedorova, N.; Lin, D.; Hari, K.L.; Jain, R.; Rodas, J.D.; Das, S.R.; Shabman, R.S.; Weaver, S.C. Comprehensive Genome Scale Phylogenetic Study Provides New Insights on the Global Expansion of Chikungunya Virus. J. Virol. 2016, 90, 10600–10611. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.C. Arrival of Chikungunya Virus in the New World: Prospects for Spread and Impact on Public Health. PLoS Negl. Trop. Dis. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.V.J., Jr.; Ludwig-Begall, L.F.; Oliveira-Filho, E.F.d.; Oliveira, R.A.S.; Durães-Carvalho, R.; Lopes, T.R.R.; Silva, D.E.A.; Gil, L.H.V.G. A scoping review of Chikungunya virus infection: Epidemiology, clinical characteristics, viral co-circulation complications, and control. Acta Trop. 2018, 188, 213–224. [Google Scholar] [CrossRef]
- Nunes, M.R.T.; Faria, N.R.; de Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.G.; de Oliveira, L.F.; da Silva Azevedo, R.d.S.; da Silva, D.E.A.; da Silva, E.V.P.; da Silva, S.P.; et al. Emergence and potential for spread of Chikungunya virus in Brazil. BMC Med. 2015, 13, 102. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.G.; Andrade, A.M.S.; Da Costa, M.C.N.; Castro, J.S.M.; Oliveira, F.L.S.; Goes, C.S.B.; Maia, M.; Santana, E.B.; Nunes, B.T.D.; Vasconcelos, P.F.C. East/central/South African genotype chikungunya virus, Brazil, 2014. Emerg. Infect. Dis. 2015, 21, 906–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ministério da Saúde Ministério da Saúde, 2020. Monitoramento dos casos de arboviroses urbanas transmitidas pelo Aedes (dengue, chikungunya e Zika), Semanas Epidemiológicas 01 a 52. Available online: https://www.saude.gov.br/images/pdf/2020/marco/06/Boletim-epidemiologico-SVS-10.pdf (accessed on 8 May 2020).
- Mayer, S.V.; Tesh, R.B.; Vasilakis, N. The emergence of arthropod-borne viral diseases: A global prospective on dengue, chikungunya and zika fevers. Acta Trop. 2017, 166, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Izurieta, R.O.; DeLacure, D.A.; Izurieta, A.; Hoare, I.A.; Reina Ortiz, M. Mayaro virus: The jungle flu. Virus Adapt. Treat. 2018, Volume 10, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Gould, E.; Pettersson, J.; Higgs, S.; Charrel, R.; de Lamballerie, X. Emerging arboviruses: Why today? One Heal. 2017, 4, 1–13. [Google Scholar] [CrossRef]
- Long, K.C.; Ziegler, S.A.; Thangamani, S.; Hausser, N.L.; Kochel, T.J.; Higgs, S.; Tesh, R.B. Experimental transmission of Mayaro virus by Aedes aegypti. Am. J. Trop. Med. Hyg. 2011, 85, 750–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiggins, K.; Eastmond, B.; Alto, B.W. Transmission potential of Mayaro virus in Florida Aedes aegypti and Aedes albopictus mosquitoes. Med. Vet. Entomol. 2018, 32, 436–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira Serra, O.; Fernandes Cardoso, B.; Maria Ribeiro, A.L.; dos Santos, F.A.L.; Dezengrini Slhessarenko, R. Mayaro virus and dengue virus 1 and 4 natural infection in culicids from Cuiabá, state of Mato Grosso, Brazil. Mem. Inst. Oswaldo Cruz 2016, 111, 20–29. [Google Scholar] [CrossRef]
- Lwande, O.W.; Obanda, V.; Lindström, A.; Ahlm, C.; Evander, M.; Näslund, J.; Bucht, G. Globe-Trotting Aedes aegypti and Aedes albopictus: Risk Factors for Arbovirus Pandemics. Vector-Borne Zoonotic Dis. 2020, 20, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Vieira, C.J.d.S.P.; Machado, L.C.; Pena, L.J.; de Morais Bronzoni, R.V.; Wallau, G.L. Spread of two Zika virus lineages in Midwest Brazil. Infect. Genet. Evol. 2019, 75, 103974. [Google Scholar] [CrossRef]
- Forrester, N.L.; Palacios, G.; Tesh, R.B.; Savji, N.; Guzman, H.; Sherman, M.; Weaver, S.C.; Lipkin, W.I. Genome-Scale Phylogeny of the Alphavirus Genus Suggests a Marine Origin. J. Virol. 2012, 86, 2729–2738. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.d.B.; Ochsenreiter, R.; Hostager, R.; Hofacker, I.L.; Janies, D.; Wolfinger, M.T. Updated phylogeny of Chikungunya virus suggests lineage-specific RNA architecture. bioRxiv 2019, 698522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Morais Bronzoni, R.V.; Baleotti, F.G.; Nogueira, M.R.R.; Nunes, M.; Figueiredo, L.T.M. Duplex Reverse Transcription-PCR Followed by Nested PCR Assays for Detection and Identification of Brazilian Alphaviruses and Flaviviruses. J. Clin. Microbiol. 2005, 43, 696–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedes, D.R.; Paiva, M.H.; Donato, M.M.; Barbosa, P.P.; Krokovsky, L.; Rocha, S.W.S.; La Saraiva, K.; Crespo, M.M.; Rezende, T.M.; Wallau, G.L.; et al. Zika virus replication in the mosquito Culex quinquefasciatus in Brazil. Emerg. Microbes Infect. 2017, 6, e69. [Google Scholar] [CrossRef] [Green Version]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Panella, A.J.; Velez, J.O.; Lambert, A.J.; Campbell, G.L. Chikungunya virus in US travelers returning from India, 2006. Emerg. Infect. Dis. 2007, 13, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Machado, L.C.; de Morais-Sobral, M.C.; Campos, T.d.L.; Pereira, M.R.; de Albuquerque, M.d.F.P.M.; Gilbert, C.; Franca, R.F.O.; Wallau, G.L. Genome sequencing reveals coinfection by multiple chikungunya virus genotypes in a recent outbreak in Brazil. PLoS Negl. Trop. Dis. 2019, 13, e0007332. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, bbx108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A. FigTree: Tree Figure Drawing Tool Version 1.4.3; Institute of Evolutionary Biology, Ed.; University of Edinburgh: Edinburgh, UK, 2014. [Google Scholar]
- Tsetsarkin, K.A.; McGee, C.E.; Volk, S.M.; Vanlandingham, D.L.; Weaver, S.C.; Higgs, S. Epistatic roles of E2 glycoprotein mutations in adaption of Chikungunya virus to Aedes albopictus and Ae. Aegypti mosquitoes. PLoS ONE 2009, 4, e90972. [Google Scholar] [CrossRef] [Green Version]
- Tsetsarkin, K.A.; Chen, R.; Leal, G.; Forrester, N.; Higgs, S.; Huang, J.; Weaver, S.C. Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes. Proc. Natl. Acad. Sci. USA 2011, 108, 7872–7877. [Google Scholar] [CrossRef] [Green Version]
- Auguste, A.J.; Liria, J.; Forrester, N.L.; Giambalvo, D.; Moncada, M.; Long, K.C.; Morón, D.; de Manzione, N.; Tesh, R.B.; Halsey, E.S.; et al. Evolutionary and ecological characterization of mayaro virus strains isolated during an outbreak, Venezuela, 2010. Emerg. Infect. Dis. 2015, 21, 1742–1750. [Google Scholar] [CrossRef]
- Pauvolid-Corrêa, A.; Soares Juliano, R.; Campos, Z.; Velez, J.; Nogueira, R.M.R.; Komar, N. Neutralising antibodies for mayaro virus in Pantanal, Brazil. Mem. Inst. Oswaldo Cruz 2015, 110, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Beckham, J.D.; Tyler, K.L. Arbovirus Infections. Contin. Lifelong Learn. Neurol. 2015, 21, 1599–1611. [Google Scholar] [CrossRef] [Green Version]
- Brazilian Ministry of Health Boletins epidemiológicos. Available online: https://www.saude.gov.br/boletins-epidemiologicos (accessed on 8 May 2020).
- Zanotto, P.M.d.A.; Leite, L.C.d.C. The Challenges Imposed by Dengue, Zika, and Chikungunya to Brazil. Front. Immunol. 2018, 9, 1964. [Google Scholar] [CrossRef]
- Secretaria de Estado de Saúde de Mato Grosso Boletim Epidemiológico: Dengue, febre de chikungunya e febre pelo vírus Zika. Available online: http://www.saude.mt.gov.br/dengue/arquivos/526/documentos (accessed on 8 May 2020).
- Moraes, M.M.; Kubiszeski, J.R.; Vieira, C.J.d.S.P.; Gusmao, A.F.; Pratis, T.S.; Colombo, T.E.; Thies, S.F.; Araujo, F.d.C.; Zanelli, C.F.; Milhim, B.H.G.d.A.; et al. Concomitant detection of Saint Louis encephalitis virus in two Brazilian States. Mem. Inst. Oswaldo Cruz. (under review).
- Mota, M.T.O.; Vedovello, D.; Estofolete, C.; Malossi, C.D.; Araújo, J.P.; Nogueira, M.L. Complete genome sequence of mayaro virus imported from the Amazon Basin to São Paulo State, Brazil. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuchi, N.; Da Silva Heinen, L.B.; Dos Santos, M.A.M.; Pereira, F.C.; Slhessarenko, R.D. Molecular detection of Mayaro virus during a dengue outbreak in the state of Mato Grosso, Central-West Brazil. Mem. Inst. Oswaldo Cruz 2014, 109, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Brunini, S.; França, D.D.S.; Silva, J.B.; Silva, L.N.; Silva, F.P.A.; Spadoni, M.; Rezza, G. High frequency of mayaro virus IgM among febrile patients, central Brazil. Emerg. Infect. Dis. 2017, 23, 1025–1026. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Chua, C.-L.; Sam, I.-C.; Chiam, C.-W.; Chan, Y.-F. The neutralizing role of IgM during early Chikungunya virus infection. PLoS ONE 2017, 12, e0171989. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, M.J.; De Souza, W.M.; Romeiro, M.F.; de Souza Costa, M.C.; Slhessarenko, R.D.; Figueiredo, L.T.M. Development of an enzyme-linked immunosorbent assay to detect antibodies targeting recombinant envelope protein 2 of Mayaro virus. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.D.; Lee, J.E. The Secret Life of Viral Entry Glycoproteins: Moonlighting in Immune Evasion. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.M.; Aguilar, P.V.; Chandler, L.J.; Brault, A.C.; Meakins, T.A.; Watts, D.; Russell, K.L.; Olson, J.; Vasconcelos, P.F.C.; Da Rosa, A.T.; et al. Genetic relationships among Mayaro and Una viruses suggest distinct patterns of transmission. Am. J. Trop. Med. Hyg. 2006, 75, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.M.; Dias, R.S.; de Oliveira, M.D.; Costa, I.C.T.A.; Fernandes, L.d.S.; Pessoa, C.R.; da Matta, S.L.P.; Costa, V.V.; Souza, D.G.; da Silva, C.C.; et al. Animal model of arthritis and myositis induced by the Mayaro virus. PLoS Negl. Trop. Dis. 2019, 13, e0007375. [Google Scholar] [CrossRef]
- Harsha, P.K.; Reddy, V.; Rao, D.; Pattabiraman, C.; Mani, R.S. Continual circulation of ECSA genotype and identification of a novel mutation I317V in the E1 gene of Chikungunya viral strains in southern India during 2015–2016. J. Med. Virol. 2020, jmv.25662. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Sharma, A.K.; Sukumaran, D.; Parida, M.; Dash, P.K. Two novel epistatic mutations (E1:K211E and E2:V264A) in structural proteins of Chikungunya virus enhance fitness in Aedes aegypti. Virology 2016, 497, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Vazeille, M.; Moutailler, S.; Coudrier, D.; Rousseaux, C.; Khun, H.; Huerre, M.; Thiria, J.; Dehecq, J.-S.; Fontenille, D.; Schuffenecker, I.; et al. Two Chikungunya Isolates from the Outbreak of La Reunion (Indian Ocean) Exhibit Different Patterns of Infection in the Mosquito, Aedes albopictus. PLoS ONE 2007, 2, e1168. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A Single Mutation in Chikungunya Virus Affects Vector Specificity and Epidemic Potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Cunze, S.; Kochmann, J.; Koch, L.K.; Klimpel, S. Niche conservatism of Aedes albopictus and Aedes aegypti-Two mosquito species with different invasion histories. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mourão, M.P.G.; Bastos, M.D.S.; De Figueiredo, R.P.; Gimaque, J.B.L.; Dos Santos Galusso, E.; Kramer, V.M.; De Oliveira, C.M.C.; Naveca, F.G.; Figueiredo, L.T.M. Mayaro fever in the city of manaus, Brazil, 2007–2008. Vector-Borne Zoonotic Dis. 2012, 12, 42–46. [Google Scholar] [CrossRef]
- Martins, K.A.; Gregory, M.K.; Valdez, S.M.; Sprague, T.R.; Encinales, L.; Pacheco, N.; Cure, C.; Porras-Ramirez, A.; Rico-Mendoza, A.; Chang, A.; et al. Neutralizing Antibodies from Convalescent Chikungunya Virus Patients Can Cross-Neutralize Mayaro and Una Viruses. Am. J. Trop. Med. Hyg. 2019, 100, 1541–1544. [Google Scholar] [CrossRef]
- Webb, E.M.; Azar, S.R.; Haller, S.L.; Langsjoen, R.M.; Cuthbert, C.E.; Ramjag, A.T.; Luo, H.; Plante, K.; Wang, T.; Simmons, G.; et al. Effects of Chikungunya virus immunity on Mayaro virus disease and epidemic potential. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Naveca, F.G.; Claro, I.; Giovanetti, M.; de Jesus, J.G.; Xavier, J.; Iani, F.C.d.M.; do Nascimento, V.A.; de Souza, V.C.; Silveira, P.P.; Lourenço, J.; et al. Genomic, epidemiological and digital surveillance of Chikungunya virus in the Brazilian Amazon. PLoS Negl. Trop. Dis. 2019, 13, e0007065. [Google Scholar] [CrossRef] [Green Version]
- Fischer, C.; de Lamballerie, X.; Drexler, J.F. Enhanced Molecular Surveillance of Chikungunya Virus. mSphere 2019, 4, e00295-19. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, L.T.M. Large outbreaks of chikungunya virus in Brazil reveal uncommon clinical features and fatalities. Rev. Soc. Bras. Med. Trop. 2017, 50, 583–584. [Google Scholar] [CrossRef] [PubMed]
- De O’Mota, M.T.; Avilla, C.M.S.; Nogueira, M.L. Mayaro virus: A neglected threat could cause the next worldwide viral epidemic. Future Virol. 2019, 14, 375–377. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate ID | Age | Gender | Onset of Symptoms—dd/mm/yyyy | Collection Date—dd/mm/yyyy | Sample | qRT-PCR Ct a | Genbank Accession Code | Total No. Reads b | No. Mapped Reads (%) b | Depth of Coverage b | Genome Length (nt) b |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H307 MAYV | 21 | F | 14/03/2015 | 16/03/2015 | C6/36 passage 1 | 6.12 | MH513597 | 2,813,153 | 2,376,618 (84.48%) | 15,453.99 | 11,147 |
| H542 CHIKV | 20 | F | 04/02/2018 | 07/02/2018 | Serum | 23.19 | MT349960 | 1,123,424 | 1,083,817 (96.47%) | 7133.15 | 11,492 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Julia da Silva Pessoa Vieira, C.; José Ferreira da Silva, D.; Rigotti Kubiszeski, J.; Ceschini Machado, L.; Pena, L.J.; Vieira de Morais Bronzoni, R.; da Luz Wallau, G. The Emergence of Chikungunya ECSA Lineage in a Mayaro Endemic Region on the Southern Border of the Amazon Forest. Trop. Med. Infect. Dis. 2020, 5, 105. https://doi.org/10.3390/tropicalmed5020105
Julia da Silva Pessoa Vieira C, José Ferreira da Silva D, Rigotti Kubiszeski J, Ceschini Machado L, Pena LJ, Vieira de Morais Bronzoni R, da Luz Wallau G. The Emergence of Chikungunya ECSA Lineage in a Mayaro Endemic Region on the Southern Border of the Amazon Forest. Tropical Medicine and Infectious Disease. 2020; 5(2):105. https://doi.org/10.3390/tropicalmed5020105
Chicago/Turabian StyleJulia da Silva Pessoa Vieira, Carla, David José Ferreira da Silva, Janaína Rigotti Kubiszeski, Laís Ceschini Machado, Lindomar José Pena, Roberta Vieira de Morais Bronzoni, and Gabriel da Luz Wallau. 2020. "The Emergence of Chikungunya ECSA Lineage in a Mayaro Endemic Region on the Southern Border of the Amazon Forest" Tropical Medicine and Infectious Disease 5, no. 2: 105. https://doi.org/10.3390/tropicalmed5020105
APA StyleJulia da Silva Pessoa Vieira, C., José Ferreira da Silva, D., Rigotti Kubiszeski, J., Ceschini Machado, L., Pena, L. J., Vieira de Morais Bronzoni, R., & da Luz Wallau, G. (2020). The Emergence of Chikungunya ECSA Lineage in a Mayaro Endemic Region on the Southern Border of the Amazon Forest. Tropical Medicine and Infectious Disease, 5(2), 105. https://doi.org/10.3390/tropicalmed5020105