
Genetic Analysis of Influenza A/H1N1pdm Strains Isolated in Bangladesh in Early 2020

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Viruses and Cells
2.3. Clinical Specimens
2.4. Virus Isolation
2.5. RT-PCR for the Classification of Influenza Virus Subtypes in the Clinical Specimens
2.6. Absolute Quantitation of Influenza Viral RNA Using the SYBR Green I Real-Time RT-PCR Assay
2.7. Sequencing of the HA and NA Genome Segments of IAV H1N1pdm09
2.8. Sequence Characterization
2.9. Phylogenetic Analysis
3. Results
3.1. Background of the Clinical Specimens, RT-PCR Analysis, and Virus Isolation
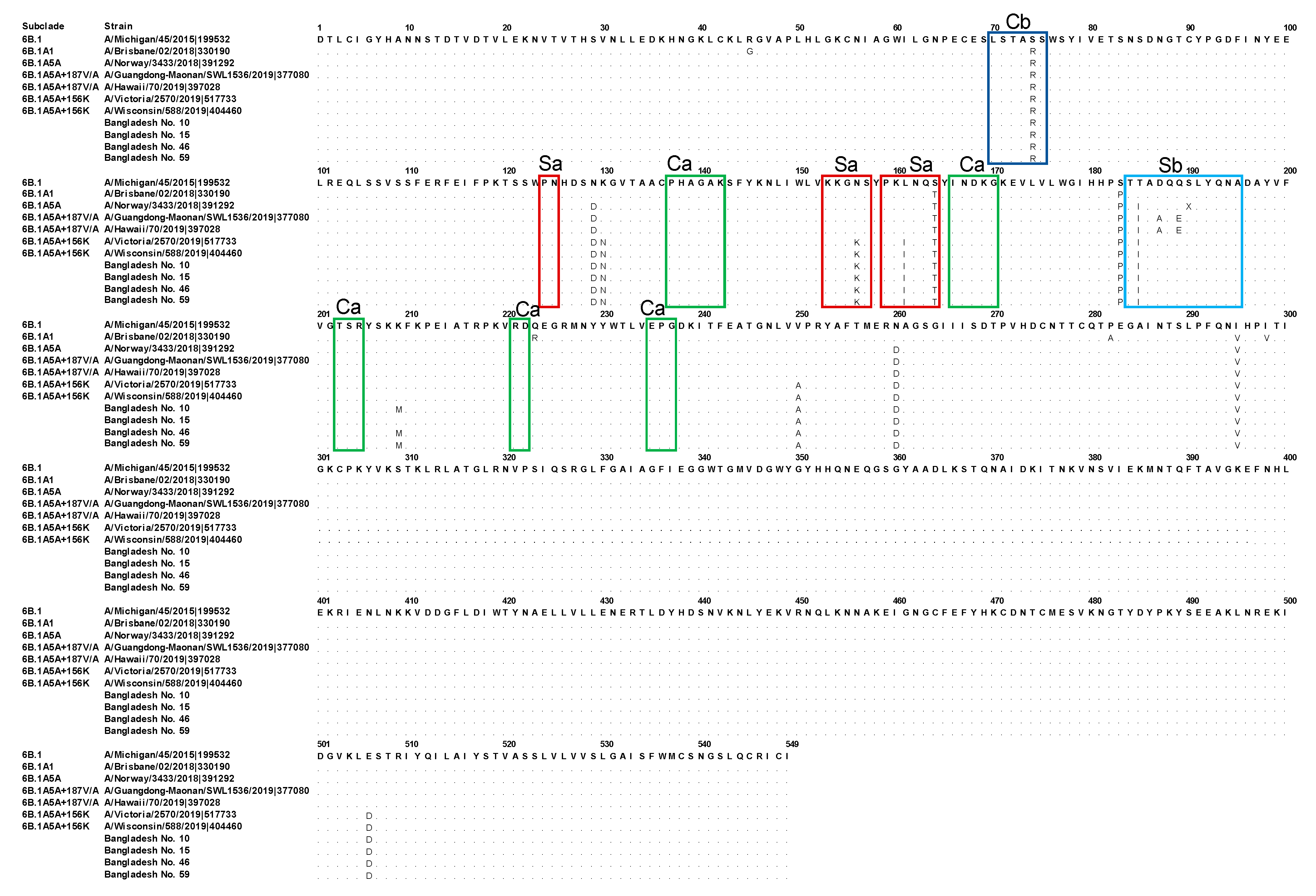
3.2. Genetic Characterization of the HA Segments Obtained in the Present Study
3.3. Human IAV H1N1pdm09 Detected in Bangladesh from 2018 to 2020
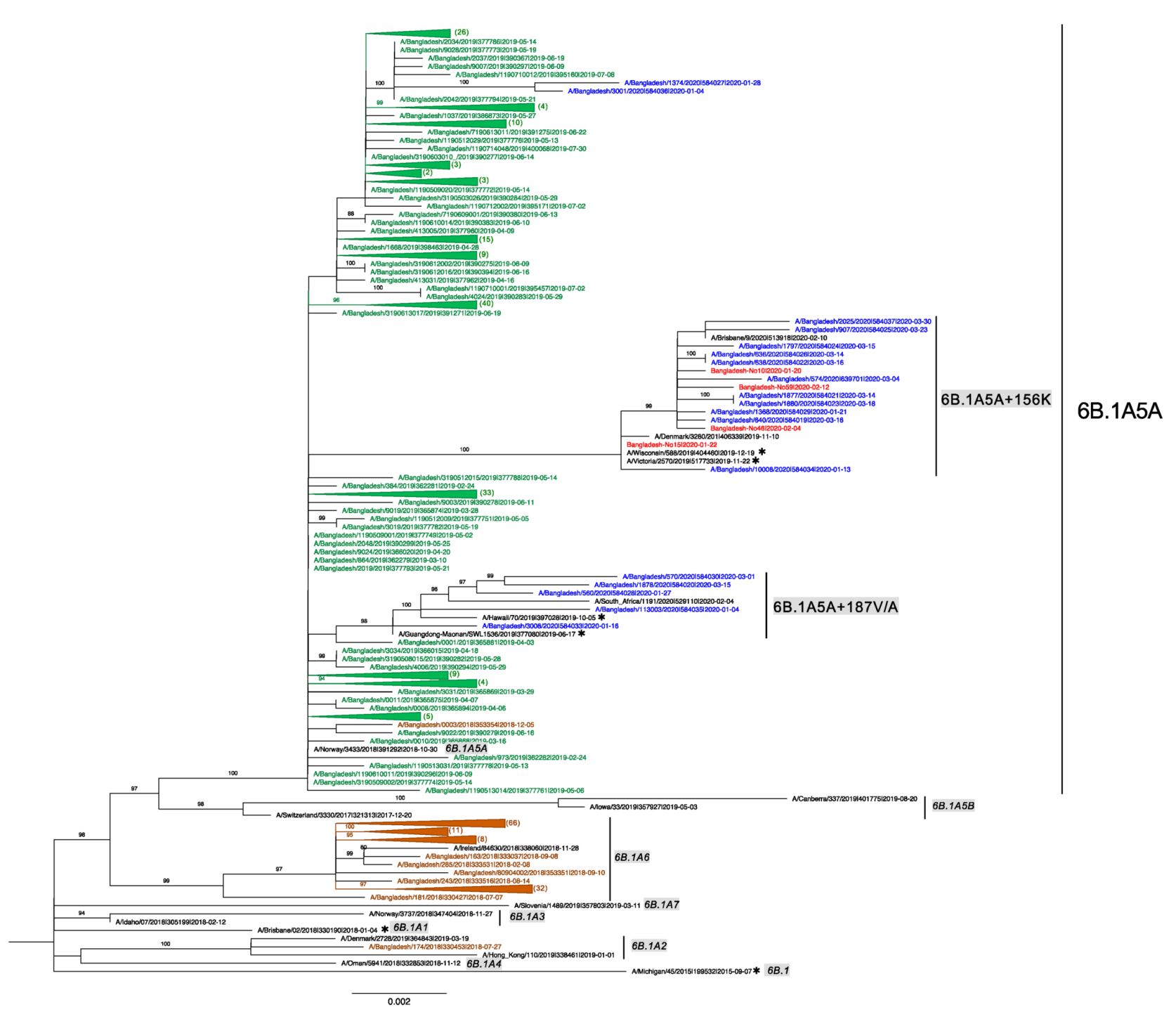
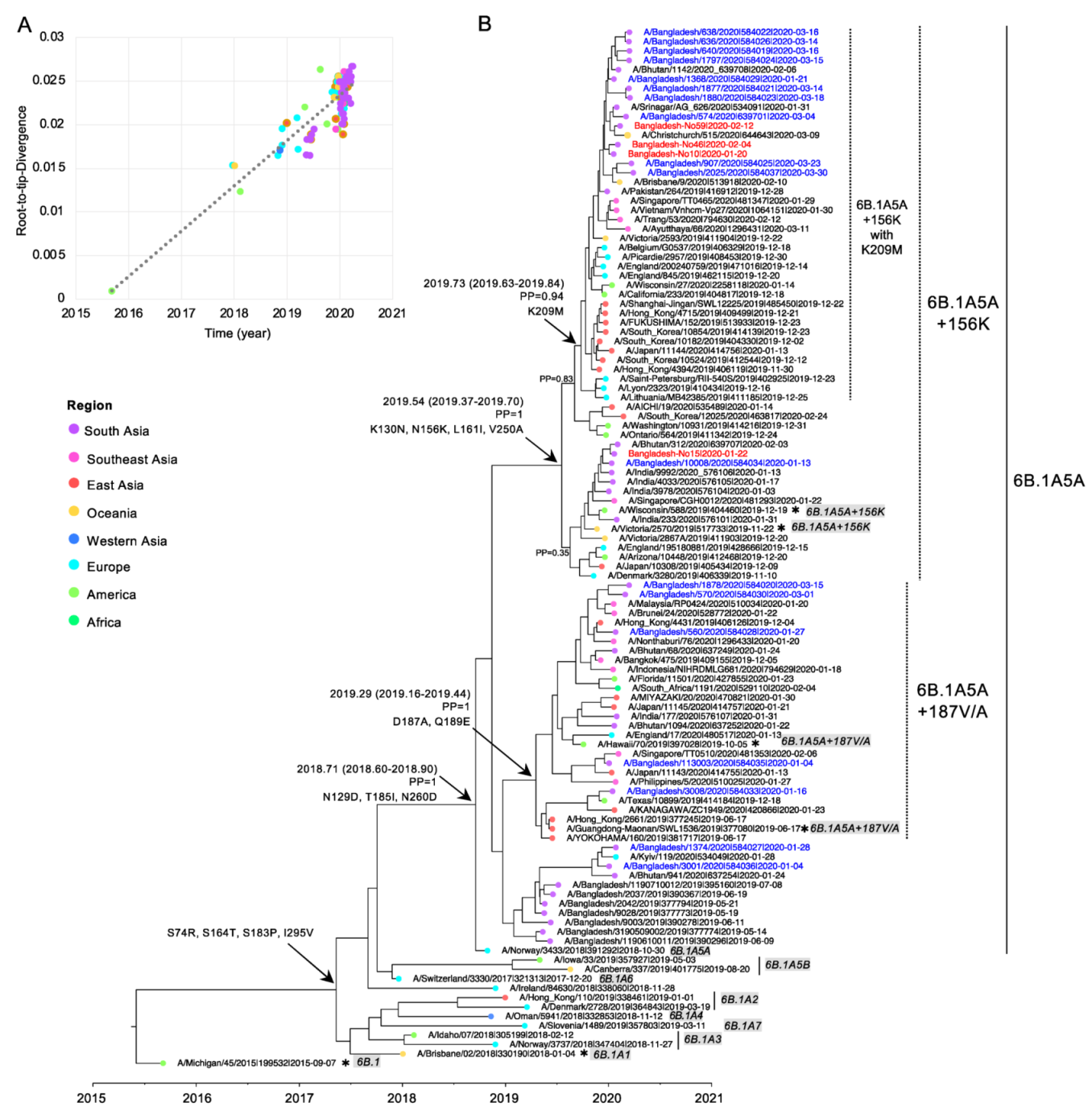
3.4. Evolution of Subclades 6B.1A5A + 187V/A and 6B.1A5A + 156K of Bangladesh 2020 Human IAV H1N1pdm09
3.5. Global Circulation of Subclades 6B.1A5A + 187A and 6B.1A5A + 156K
3.6. Genetic Characterization of NA Segments Obtained in the Present Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco-Cabrera, S.; Gomez, J.; et al. New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retamal, M.; Abed, Y.; Corbeil, J.; Boivin, G. Epitope mapping of the 2009 pandemic and the A/Brisbane/59/2007 seasonal (H1N1) influenza virus haemagglutinins using mAbs and escape mutants. J. Gen. Virol. 2014, 95, 2377–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Ekiert, D.C.; Krause, J.C.; Hai, R.; Crowe, J.E.; Wilson, I.A. Structural Basis of Preexisting Immunity to the 2009 H1N1 Pandemic Influenza Virus. Science 2010, 328, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.; Corper, A.L.; Basler, C.F.; Taubenberger, J.K.; Palese, P.; Wilson, I.A. Structure of the Uncleaved Human H1 Hemagglutinin from the Extinct 1918 Influenza Virus. Science 2004, 303, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Borkenhagen, L.K.; Salman, M.D.; Ma, M.-J.; Gray, G.C. Animal influenza virus infections in humans: A commentary. Int. J. Infect. Dis. 2019, 88, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.I.; Holmes, E. The evolution of epidemic influenza. Nat. Rev. Genet. 2007, 8, 196–205. [Google Scholar] [CrossRef]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treanor, J. Influenza Vaccine—Outmaneuvering Antigenic Shift and Drift. N. Engl. J. Med. 2004, 350, 218–220. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Moazzem, K.; Chowdhury, M.; Sehrin, F. Connecting South Asia and Southeast Asia: A Bangladesh Country Study; ADBI Working Paper No. 500; Asian Development Bank Institute: Tokyo, Japan, 2014. [Google Scholar]
- Sasaki, T.; Kubota-Koketsu, R.; Takei, M.; Hagihara, T.; Iwamoto, S.; Murao, T.; Sawami, K.; Fukae, D.; Nakamura, M.; Nagata, E.; et al. Reliability of a Newly-Developed Immunochromatography Diagnostic Kit for Pandemic Influenza A/H1N1pdm Virus: Implications for Drug Administration. PLoS ONE 2012, 7, e50670. [Google Scholar]
- Karlsson, M.; Wallensten, A.; Lundkvist, Å.; Olsen, B.; Brytting, M. A real-time PCR assay for the monitoring of influenza a virus in wild birds. J. Virol. Methods 2007, 144, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.-M.; Spirason, N.; Iannello, P.; Jelley, L.; Lau, H.; Barr, I.G. A simplified Sanger sequencing method for full genome sequencing of multiple subtypes of human influenza A viruses. J. Clin. Virol. 2015, 68, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; Von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Baillie, G.J.; Galiano, M.; Agapow, P.M.; Myers, R.; Chiam, R.; Gall, A.; Palser, A.L.; Watson, S.J.; Hedge, J.; Underwood, A.; et al. Evolutionary dynamics of local pandemic H1N1/2009 influenza virus lineages revealed by whole-genome analysis. J. Virol. 2012, 86, 11–18. [Google Scholar] [CrossRef] [Green Version]
- ECDC. Influenza Virus Characterisation Summary Europe, Febuary 2021; Surveillance Report; ECDC: Solna Municipality, Sweden, 2021. [Google Scholar]
- WHO. Recommended Composition of Influenza Virus Vaccines for Use in the 2021–2022 Northern Hemisphere Influenza Season; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Pinilla, L.T.; Holder, B.P.; Abed, Y.; Boivin, G.; Beauchemin, C. The H275Y Neuraminidase Mutation of the Pandemic A/H1N1 Influenza Virus Lengthens the Eclipse Phase and Reduces Viral Output of Infected Cells, Potentially Compromising Fitness in Ferrets. J. Virol. 2012, 86, 10651–10660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engebretsen, S.; Engø-Monsen, K.; Aleem, M.A.; Gurley, E.S.; Frigessi, A.; de Blasio, B.F. Time-aggregated mobile phone mobility data are sufficient for modelling influenza spread: The case of Bangladesh. J. R. Soc. Interface 2020, 17, 20190809. [Google Scholar] [CrossRef] [PubMed]
- Strengell, M.; Ikonen, N.; Ziegler, T.; Julkunen, I. Minor Changes in the Hemagglutinin of Influenza A(H1N1)2009 Virus Alter Its Antigenic Properties. PLoS ONE 2011, 6, e25848. [Google Scholar] [CrossRef] [Green Version]
- Guarnaccia, T.; Carolan, L.A.; Maurer-Stroh, S.; Lee, R.T.C.; Job, E.; Reading, P.; Petrie, S.; McCaw, J.; McVernon, J.; Hurt, A.; et al. Antigenic Drift of the Pandemic 2009 A(H1N1) Influenza Virus in a Ferret Model. PLoS Pathog. 2013, 9, e1003354. [Google Scholar] [CrossRef] [Green Version]
- Le Nguyen, H.K.; Nguyen, P.T.K.; Nguyen, T.C.; Hoang, P.V.M.; Le, T.T.; Vuong, C.D.; Nguyen, A.P.; Tran, L.T.T.; Nguyen, B.G.; Lê, M.Q. Virological characterization of influenza H1N1pdm09 in Vietnam, 2010–2013. Influenza Respir. Viruses 2015, 9, 216–224. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Kondor, R.J.G.; Chung, J.R.; Zimmerman, R.K.; Nowalk, M.P.; Jackson, M.L.; Jackson, L.A.; Monto, A.S.; Martin, E.T.; Belongia, E.A.; et al. Effect of Antigenic Drift on Influenza Vaccine Effectiveness in the United States—2019–2020. Clin. Infect. Dis. 2020, 73, e4244–e4250. [Google Scholar] [CrossRef]
- WHO. Recommended Composition of Influenza Virus Vaccines for Use in the 2021 Southern Hemisphere Influenza Season; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Bénet, T.; Amour, S.; Valette, M.; Saadatian-Elahi, M.; Aho-Glélé, L.S.; Berthelot, P.; Denis, M.-A.; Grando, J.; Landelle, C.; Astruc, K.; et al. Incidence of Asymptomatic and Symptomatic Influenza Among Healthcare Workers: A Multicenter Prospective Cohort Study. Clin. Infect. Dis. 2020, 72, e311–e318. [Google Scholar] [CrossRef]
- Patrozou, E.; Mermel, L.A. Does influenza transmission occur from asymptomatic infection or prior to symptom onset? Public Health Rep. 2009, 124, 193–196. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, Z.-H.; Ye, L.-L.; He, X.-L.; Wei, X.-S.; Ma, Y.-L.; Li, H.; Chen, L.; Wang, X.-R.; Zhou, Q. Co-infection of SARS-CoV-2 and Influenza A Virus: A Case Series and Fast Review. Curr. Med. Sci. 2021, 41, 51–57. [Google Scholar] [CrossRef]
- Zhao, X.-N.; Zhang, H.-J.; Li, D.; Zhou, J.-N.; Chen, Y.-Y.; Sun, Y.-H.; Adeola, A.C.; Fu, X.-Q.; Shao, Y.; Zhang, M.-L. Whole-genome sequencing reveals origin and evolution of influenza A(H1N1)pdm09 viruses in Lincang, China, from 2014 to 2018. PLoS ONE 2020, 15, e0234869. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, A.; Sasaki, T.; Phadungsombat, J.; Koketsu, R.; Rahim, R.; Ara, N.; Biswas, S.M.; Yonezawa, R.; Nakayama, E.E.; Rahman, M.; et al. Genetic Analysis of Influenza A/H1N1pdm Strains Isolated in Bangladesh in Early 2020. Trop. Med. Infect. Dis. 2022, 7, 38. https://doi.org/10.3390/tropicalmed7030038
Hasan A, Sasaki T, Phadungsombat J, Koketsu R, Rahim R, Ara N, Biswas SM, Yonezawa R, Nakayama EE, Rahman M, et al. Genetic Analysis of Influenza A/H1N1pdm Strains Isolated in Bangladesh in Early 2020. Tropical Medicine and Infectious Disease. 2022; 7(3):38. https://doi.org/10.3390/tropicalmed7030038
Chicago/Turabian StyleHasan, Abu, Tadahiro Sasaki, Juthamas Phadungsombat, Ritsuko Koketsu, Rummana Rahim, Nikhat Ara, Suma Mita Biswas, Riku Yonezawa, Emi E. Nakayama, Mizanur Rahman, and et al. 2022. "Genetic Analysis of Influenza A/H1N1pdm Strains Isolated in Bangladesh in Early 2020" Tropical Medicine and Infectious Disease 7, no. 3: 38. https://doi.org/10.3390/tropicalmed7030038
APA StyleHasan, A., Sasaki, T., Phadungsombat, J., Koketsu, R., Rahim, R., Ara, N., Biswas, S. M., Yonezawa, R., Nakayama, E. E., Rahman, M., & Shioda, T. (2022). Genetic Analysis of Influenza A/H1N1pdm Strains Isolated in Bangladesh in Early 2020. Tropical Medicine and Infectious Disease, 7(3), 38. https://doi.org/10.3390/tropicalmed7030038