

In Silico Approaches for the Identification of Aptamer Binding Interactions to Leptospira spp. Cell Surface Proteins



Abstract
:1. Introduction
2. Materials and Methods
2.1. Aptamers and Leptospira spp. Outer Membrane Protein Targets
2.2. Software Workflow
2.3. Validation of Aptamer-Target Binding
3. Results
3.1. Aptamer–Target Proteins and Their Predicted Structures
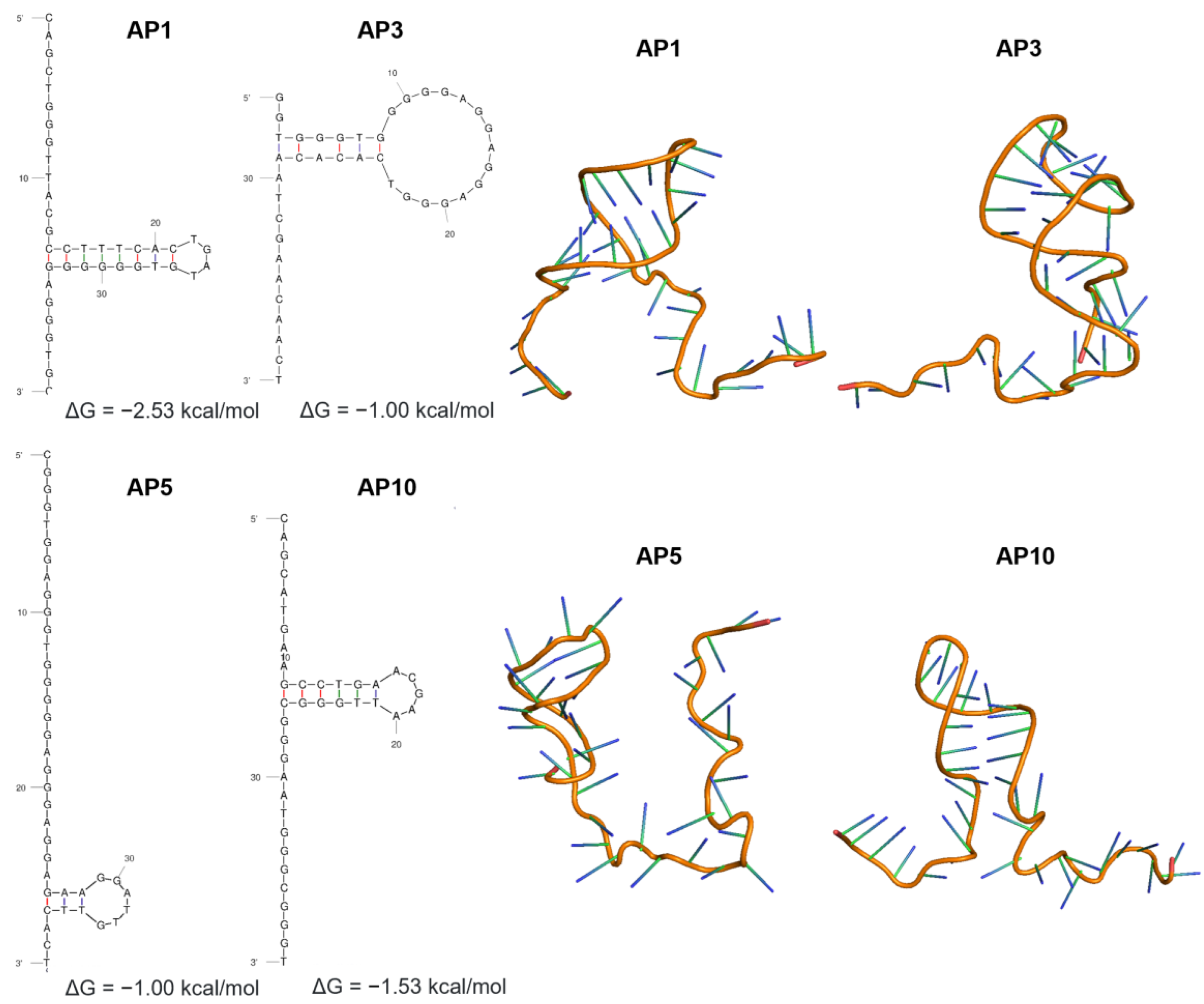
3.2. Secondary and Tertiary Structures of Candidate Aptamers
3.3. Evaluation of the Binding Capacity of Aptamers by Direct Enzyme-Linked Aptamer Assay (ELAA)
3.4. Aptamer and Leptospira Cell Surface Protein Complexes and Their Interacting Residues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar Kulabhusan, P.; Hussain, B.; Yüce, M. Current perspectives on aptamers as diagnostic tools and therapeutic agents. Pharmaceutics 2020, 12, 646. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Mosing, R.K.; Bowser, M.T. Isolating aptamers using capillary electrophoresis-SELEX (CE-SELEX). Methods Mol. Biol. Clifton NJ 2009, 535, 33–43. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, W.; Chen, S.; Zhuang, Z.; Zhang, Y.; Jiang, L.; LIN, J.S. SELEX tool: A novel and convenient gel-based diffusion method for monitoring of aptamer-target binding. J. Biol. Eng. 2020, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Li, H.; Sun, W.; Xu, D.; He, F. Rapid selection of aptamers based on protein microarray. RSC Adv. 2019, 9, 9762–9768. [Google Scholar] [CrossRef] [Green Version]
- Zulkeflee Sabri, M.; Azzar Abdul Hamid, A.; Mariam Sayed Hitam, S.; Zulkhairi Abdul Rahim, M. In-silico selection of aptamer: A review on the revolutionary approach to understand the aptamer design and interaction through computational chemistry. Mater Today Proc. 2019, 19, 1572–1581. [Google Scholar] [CrossRef]
- Chushak, Y.; Stone, M.O. In silico selection of RNA aptamers. Nucleic Acids Res. 2009, 37, e87. [Google Scholar] [CrossRef] [Green Version]
- Kinghorn, A.B.; Fraser, L.A.; Liang, S.; Shiu, S.C.; Tanner, J.A. Aptamer Bioinformatics. Int. J. Mol. Sci. 2017, 18, 2516. [Google Scholar] [CrossRef] [Green Version]
- Navien, T.N.; Thevendran, R.; Hamdani, H.Y.; Tang, T.-H.; Citartan, M. In silico molecular docking in DNA aptamer development. Biochimie 2021, 180, 54–67. [Google Scholar] [CrossRef]
- Bell, D.R.; Weber, J.K.; Yin, W.; Huynh, T.; Duan, W.; Zhou, R. Ruhong In silico design and validation of high-affinity RNA aptamers targeting epithelial cellular adhesion molecule dimers. Proc. Natl. Acad. Sci. USA 2020, 117, 8486–8493. [Google Scholar] [CrossRef]
- Naing, C.; Reid, S.A.; Aye, S.N.; Htet, N.H.; Ambu, S. Risk factors for human leptospirosis following flooding: A meta-analysis of observational studies. PLoS ONE 2019, 14, e0217643. [Google Scholar] [CrossRef]
- Chacko, C.S.; Lakshmi, S.S.; Jayakumar, A.; Binu, S.L.; Pant, R.D.; Giri, A.; Chand, S.; UP, N. A short review on leptospirosis: Clinical manifestations, diagnosis and treatment. Clin. Epidemiol. Glob. Health 2021, 11, 100741. [Google Scholar] [CrossRef]
- Oliveira, R.; Pinho, E.; Sousa, A.L.; Dias, Ó.; Azevedo, N.F.; Almeida, C. Modelling aptamers with nucleic acid mimics (NAM): From sequence to three-dimensional docking. PLoS ONE 2022, 17, e0264701. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Huang, Y.; Xiao, Y. 3dRNA v2.0: An updated web server for RNA 3D structure prediction. Int. J. Mol. Sci. 2019, 20, 4116. [Google Scholar] [CrossRef] [Green Version]
- Dassault Systèmes BIOVIA 2020. Available online: https://biovia-discovery-studio-2020-client.software.informer.com/ (accessed on 4 July 2022).
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Podgoršek, D.; Ružić-Sabljić, E.; Logar, M.; Pavlović, A.; Remec, T.; Baklan, Z.; Pal, E.; Cerar, T. Evaluation of real-time PCR targeting the lipL32 gene for diagnosis of Leptospira infection. BMC Microbiol. 2020, 20, 59. [Google Scholar] [CrossRef]
- Hauk, P.; Negrotto, S.; Romero, E.C.; Vasconcellos, S.A.; Genovez, M.É.; Ward, R.J.; Schattner, M.; Goméz, R.M.; Ho, P.L. Expression and characterization of HlyX hemolysin from Leptospira interrogans serovar Copenhageni: Potentiation of hemolytic activity by LipL32. Biochem. Biophys. Res. Commun. 2005, 333, 1341–1347. [Google Scholar] [CrossRef]
- Haake, D.A.; Zückert, W.R. The leptospiral outer membrane. Curr. Top. Microbiol. Immunol. 2015, 387, 187–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maia, M.A.C.; Bettin, E.B.; Barbosa, L.N.; de Oliveira, N.R.; Bunde, T.T.; Pedra, A.C.K.; Rosa, G.A.; da Rosa, E.E.B.; Seixas Neto, A.C.P.; Grassmann, A.A.; et al. Challenges for the development of a universal vaccine against leptospirosis revealed by the evaluation of 22 vaccine candidates. Front. Cell. Infect. Microbiol. 2022, 12, 940966. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, K.V.; Lourdault, K.; Matsunaga, J.; Haake, D.A. Immunoprotective properties of recombinant LigA and LigB in a hamster model of acute leptospirosis. PLoS ONE 2017, 12, e0180004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, A.; Premlatha, M.M.; Raja, V.; Akino Mercy, C.S.; Sumathi, K.; Shariff, M.; Natarajaseenivasan, K. Protective immunity of recombinant LipL21 and I-LipL21 against Leptospira interrogans serovar Autumnalis N2 infection. J. Infect. Dev. Ctries. 2018, 12, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Grassmann, A.A.; Félix, S.R.; dos Santos, C.X.; Amaral, M.G.; Seixas Neto, A.C.P.; Fagundes, M.Q.; Seixas, F.K.; da Silva, E.F.; Conceição, F.R.; Dellagostin, O.A. Protection against lethal leptospirosis after vaccination with LipL32 coupled or coadministered with the B subunit of Escherichia coli heat-labile enterotoxin. Clin. Vaccine Immunol. CVI 2012, 19, 740–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.-H.; Chang, Y.-C.; Hsiao, C.-D.; Huang, S.-H.; Wang, M.-S.; Ko, Y.-C.; Yang, C.-W.; Sun, Y.-J. LipL41, a Hemin Binding Protein from Leptospira santarosai serovar Shermani. PLoS ONE 2013, 8, e83246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, Y.; Du, Z.; Xin, X.; Ye, Q.; Xu, Y. Comparative proteomic analysis of Leptospira interrogans serogroup Icterohaemorrhagiae human vaccine strain and epidemic isolate from China. Arch. Microbiol. 2022, 204, 460. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-H.; Yang, C.-W. Insight into the structure, functions, and dynamics of the Leptospira outer membrane proteins with the pathogenicity. Membranes 2022, 12, 300. [Google Scholar] [CrossRef]
- Fernandes, L.G.V.; Vieira, M.L.; Kirchgatter, K.; Alves, I.J.; de Morais, Z.M.; Vasconcellos, S.A.; Romero, E.C.; Nascimento, A.L.T.O. OmpL1 is an extracellular matrix- and plasminogen-interacting protein of Leptospira spp. Infect. Immun. 2012, 80, 3679–3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassmann, A.A.; Kremer, F.S.; dos Santos, J.C.; Souza, J.D.; Pinto, L.D.S.; McBride, A.J.A. Discovery of novel leptospirosis vaccine candidates using reverse and structural vaccinology. Front. Immunol. 2017, 8, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buglak, A.A.; Samokhvalov, A.V.; Zherdev, A.V.; Dzantiev, B.B. Methods and applications of in silico aptamer design and modeling. Int. J. Mol. Sci. 2020, 21, 8420. [Google Scholar] [CrossRef]
- Techawiwattanaboon, T.; Thaibankluay, P.; Kreangkaiwal, C.; Sathean-Anan-Kun, S.; Khaenam, P.; Makjaroen, J.; Pisitkun, T.; Patarakul, K. Surface proteomics and label-free quantification of Leptospira interrogans serovar Pomona. PLoS Negl. Trop. Dis. 2021, 15, e0009983. [Google Scholar] [CrossRef] [PubMed]
- Cullen, P.A.; Xu, X.; Matsunaga, J.; Sanchez, Y.; Ko, A.I.; Haake, D.A.; Adler, B. Surfaceome of Leptospira spp. Infect. Immun. 2005, 73, 4853–4863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, S.-H.; Hung, C.-C.; Chang, M.-Y.; Ko, Y.-C.; Yang, H.-Y.; Hsu, H.-H.; Tian, Y.-C.; Chou, L.-F.; Pan, R.-L.; Tseng, F.-G.; et al. Active Components of Leptospira outer membrane protein LipL32 to toll-like receptor 2. Sci. Rep. 2017, 7, 8363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MB, T.; AF, T.; ALTO, N. The leptospiral LipL21 and LipL41 proteins exhibit a broad spectrum of interactions with host cell components. Virulence 2021, 12, 2798–2813. [Google Scholar] [CrossRef]
- Zhang, K.; Murray, G.L.; Seemann, T.; Srikram, A.; Bartpho, T.; Sermswan, R.W.; Adler, B.; Hoke, D.E. Leptospiral LruA is required for virulence and modulates an interaction with mammalian apolipoprotein AI. Infect. Immun. 2013, 81, 3872–3879. [Google Scholar] [CrossRef] [Green Version]
- Dellagostin, O.A.; Grassmann, A.A.; Rizzi, C.; Schuch, R.A.; Jorge, S.; Oliveira, T.L.; McBride, A.J.A.; Hartwig, D.D. Reverse vaccinology: An approach for identifying leptospiral vaccine candidates. Int. J. Mol. Sci. 2017, 18, 158. [Google Scholar] [CrossRef] [Green Version]
- Bashiru, G.; Bahaman, A.R. Advances & challenges in leptospiral vaccine development. Indian J. Med. Res. 2018, 147, 15–22. [Google Scholar] [CrossRef]
- Hoinka, J.; Zotenko, E.; Friedman, A.; Sauna, Z.E.; Przytycka, T.M. Identification of sequence-structure RNA binding motifs for SELEX-derived aptamers. Bioinformatics 2012, 28, i215–i223. [Google Scholar] [CrossRef] [Green Version]
- Elskens, J.P.; Elskens, J.M.; Madder, A. chemical modification of aptamers for increased binding affinity in diagnostic applications: Current status and future prospects. Int. J. Mol. Sci. 2020, 21, 4522. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, A.D.; Davies, D.R.; Janjic, N. Embracing proteins: Structural themes in aptamer–protein complexes. Fold. Bind. • Nucleic Acids-Protein Complexes 2016, 36, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeddi, I.; Saiz, L. Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, A.; Zimbres, F.M.; Kronenberger, T.; Wrenger, C. Molecular modeling applied to nucleic acid-based molecule development. Biomolecules 2018, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.; Yan, J.; Xiong, H.; Liu, Y.; Peng, D.; Liu, Z. Investigations on the interface of nucleic acid aptamers and binding targets. Analyst 2018, 143, 5317–5338. [Google Scholar] [CrossRef]
- Mairal, T.; Ozalp, V.C.; Lozano Sánchez, P.; Mir, M.; Katakis, I.; O’Sullivan, C.K. Aptamers: Molecular tools for analytical applications. Anal. Bioanal. Chem. 2008, 390, 989–1007. [Google Scholar] [CrossRef]
- Ahmad, N.A.; Mohamed Zulkifli, R.; Hussin, H.; Nadri, M.H. In silico approach for post-SELEX DNA aptamers: A mini-review. J. Mol. Graph. Model. 2021, 105, 107872. [Google Scholar] [CrossRef]
- Davies, D.R.; Gelinas, A.D.; Zhang, C.; Rohloff, J.C.; Carter, J.D.; O’Connell, D.; Waugh, S.M.; Wolk, S.K.; Mayfield, W.S.; Burgin, A.B.; et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. USA 2012, 109, 19971–19976. [Google Scholar] [CrossRef] [Green Version]
- Vivian, J.P.; Beddoe, T.; McAlister, A.D.; Wilce, M.C.J.; Zaker-Tabrizi, L.; Troy, S.; Byres, E.; Hoke, D.E.; Cullen, P.A.; Lo, M.; et al. Crystal structure of LipL32, the most abundant surface protein of pathogenic Leptospira spp. J. Mol. Biol. 2009, 387, 1229–1238. [Google Scholar] [CrossRef]
- Rangnekar, A.; Nash, J.A.; Goodfred, B.; Yingling, Y.G.; LaBean, T.H. Design of potent and controllable anticoagulants using DNA aptamers and nanostructures. Molecules 2016, 21, 202. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Song, J.; Wei, X.; Huang, M.; Sun, M.; Zhu, L.; Lin, B.; Shen, H.; Zhu, X.; Yang, C. Discovery of aptamers targeting the receptor-binding domain of the SARS-CoV-2 spike glycoprotein. Anal. Chem. 2020, 21, 9895–9900. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Protein Target | Function | UniProt ID | PDB File Available? | Used for Vaccine or Diagnostics |
|---|---|---|---|---|
| HlyX | Hemolysis [22] | Q72VH2 | No | No |
| LIC12575 | Outer membrane efflux protein [23,24] | Q72PA0 | No | Yes |
| LIC20151 | TonB-dependent outer membrane receptor [23,24] | Q75FN1 | No | Yes |
| LigA | Immune response [25] | G1UB65 | No | Yes |
| LipL21 | Virulence [26] | Q72WC6 | No | Yes |
| LipL32 | Inflammatory response [27] | Q72SM7 | Yes | Yes |
| LipL41 | Virulence [28,29] | Q72N71 | No | Yes |
| LipL71 | Virulence [29] | Q72TL5 | No | Yes |
| loa22 | Virulence [30] | Q7X5A5 | No | Yes |
| ompL1 | Outer membrane protein [29,31] | Q72TP4 | No | Yes |
| smc | Outer membrane receptor [32] | Q72UE3 | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almazar, C.A.; Mendoza, M.V.; Rivera, W.L. In Silico Approaches for the Identification of Aptamer Binding Interactions to Leptospira spp. Cell Surface Proteins. Trop. Med. Infect. Dis. 2023, 8, 125. https://doi.org/10.3390/tropicalmed8020125
Almazar CA, Mendoza MV, Rivera WL. In Silico Approaches for the Identification of Aptamer Binding Interactions to Leptospira spp. Cell Surface Proteins. Tropical Medicine and Infectious Disease. 2023; 8(2):125. https://doi.org/10.3390/tropicalmed8020125
Chicago/Turabian StyleAlmazar, Chembie A., Marjo V. Mendoza, and Windell L. Rivera. 2023. "In Silico Approaches for the Identification of Aptamer Binding Interactions to Leptospira spp. Cell Surface Proteins" Tropical Medicine and Infectious Disease 8, no. 2: 125. https://doi.org/10.3390/tropicalmed8020125
APA StyleAlmazar, C. A., Mendoza, M. V., & Rivera, W. L. (2023). In Silico Approaches for the Identification of Aptamer Binding Interactions to Leptospira spp. Cell Surface Proteins. Tropical Medicine and Infectious Disease, 8(2), 125. https://doi.org/10.3390/tropicalmed8020125