
Nanocellulose-Based Thermoplastic Polyurethane Biocomposites with Shape Memory Effect



Abstract
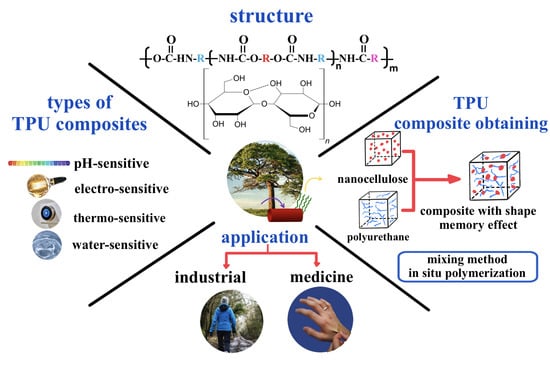
:
1. Introduction
- influence of nanocellulose on the mechanical properties of thermoplastic elastomers.
- nanocellulose in drug delivery systems [37].
2. From Cellulose to Nanocellulose
2.1. Structural Organization of Cellulose
2.2. Morphology and Methods of Isolation of Nanocellulose
2.2.1. Nanocellulose Nomenclature
- Cellulose MicroCrystals (CMCs) (e.g., MicroCrystalline Cellulose or MCC) which are a type of cellulose nanostructured material and are approximately 10–15 μm in diameter, contain mostly crystalline regions, and are composed of aggregated bundles of cellulose chains;
- Cellulose NanoCrystals (CNCs), so-called crystalline cellulose fibers [53], which are mentioned in the literature with different names, including the following:
- o
- cellulose nanowhiskers (CNWs),
- o
- cellulose crystallites,
- o
- nanorods,
- o
- nanocrystalline cellulose (NCC),
- o
- rod-like cellulose crystals.
- Cellulose nanofibrils (CNFs), so-called semicrystalline cellulose fibers, are mentioned in the literature as [29]:
- o
- nanofibrillated cellulose (NFC),
- o
- nanosized fibrillated cellulose,
- o
- cellulose fibrillar fibers,
- o
- nanofibers,
- o
- cellulose nanofibers,
- o
- aggregates of fibrils, and sometimes
- o
- CMFs or MFC.
- Bacterial nanocellulose (BNC), bacterial cellulose nanocrystals (BCNCs) or bacterial cellulose nanofibrils (BCNFs) are another type of nanocellulose that differ from cellulose nanocrystals and cellulose nanofibrils [64]. In the literature, BNC are referred to as either of the following:
- o
- Bacterial Cellulose (BC) or
- o
- Microbial Cellulose (MC).
2.2.2. Methods of Nanocellulose Extraction
- Advantec grade №1 filter paper, which consists of 100% alpha-cotton cellulose (cotton powders with a particle size of 20 mm, Sigmacell Cellulose Type 20, crystallinity 79%, and an average molecular weight of about 40,000 Da) [84].
- CMCs from Avicel in two main types depending on the CMCs particle size: Avicel PH101 (particle size less than 50 µm) and Avicel PH301 (particle size ~180 µm) [85]. This is most commonly used to obtain cellulose nanocrystals in the development of TPU nanocomposites [86]. It is purified, partially depolymerized cellulose, extracted by acid hydrolysis (chemically active extrusion) of alpha-cotton cellulose, followed by drying and milling [87].
3. Enhancing the Mechanical Properties and Shape Memory Effect of Thermoplastic Polyurethanes with Nanocellulose
3.1. Structure of Thermoplastic Polyurethanes
3.2. Influence of the Mixing Method on the Properties of Nanocellulose Enriched Materials
3.2.1. Solution Method
3.2.2. Nanocellulose Surface Modification for the Production of Polyurethane Nanocomposites with Shape Memory Effect
- Use of a CNC suspension with surface-active polysaccharides such as chitosan [24].
3.2.3. Sol–Gel Solvent Exchange Process
3.2.4. Mixing in the Melt
3.2.5. In Situ Polymerization Method
4. Hybrid Thermoplastic Polyurethane Nanocomposites Containing Nanocellulose
5. Thermoplastic Polyurethanes with Nanocellulose as Shape Memory Switching Element
5.1. Water-Sensitive Thermoplastic Polyurethane Nanocomposites Containing Nanocellulose
5.2. pH-Sensitive Thermoplastic Polyurethane Composites Containing Nanocellulose
6. Application of Composites Based on Thermoplastic Polyurethane and Nanocellulose
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baek, S.H.; Kim, J.H. Shape Memory Characteristics of Thermadapt Polyurethane Incorporated with Two Structurally Distinctive Aliphatic Isocyanates. Polym. Test. 2021, 103, 107366. [Google Scholar] [CrossRef]
- Mehrbakhsh, E.; Rezaei, M.; Babaie, A.; Mohammadi, A.; Mayan Sofla, R.L. Physical and Thermo-Mechanical Properties of Shape Memory Polyurethane Containing Reversible Chemical Cross-Links. J. Mech. Behav. Biomed. Mater. 2021, 116, 104336–104347. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S. Temperature Responsive Shape Memory Polyurethanes. Polym. Technol. Mater. 2021, 60, 1491–1518. [Google Scholar] [CrossRef]
- Constante, G.; Apsite, I.; Auerbach, P.; Aland, S.; Schönfeld, D.; Pretsch, T.; Milkin, P.; Ionov, L. Smart Mechanically Tunable Surfaces with Shape Memory Behavior and Wetting-Programmable Topography. ACS Appl. Mater. Interfaces 2022, 14, 20208–20219. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Zhang, H.; Qu, J.; Wang, J.; Bai, Y.; Sai, F. Recyclable Shape-Memory Waterborne Polyurethane Films Based on Perylene Bisimide Modified Polycaprolactone Diol. Polymers 2021, 13, 1755. [Google Scholar] [CrossRef]
- Kim, B.K.; Shin, Y.J.; Cho, S.M.; Jeong, H.M. Shape-Memory Behavior of Segmented Polyurethanes with an Amorphous Reversible Phase: The Effect of Block Length and Content. J. Polym. Sci. Part B Polym. Phys. 2000, 38, 2652–2657. [Google Scholar] [CrossRef]
- Karasu, F.; Weder, C. Tuning the Properties of Shape-Memory Polyurethanes via the Nature of the Polyester Switching Segment. Macromol. Mater. Eng. 2021, 306, 2000770–2000781. [Google Scholar] [CrossRef]
- Huang, Z.; Ban, J.; Pan, L.; Cai, S.; Liao, J. New Star-Shape Memory Polyurethanes Capable of Thermally Induced Recovery and Hydrogen Bond-Self-Healing. New J. Chem. 2021, 45, 8427–8431. [Google Scholar] [CrossRef]
- Nissenbaum, A.; Greenfeld, I.; Wagner, H.D. Shape Memory Polyurethane—Amorphous Molecular Mechanism during Fixation and Recovery. Polymer 2020, 190, 122226–122236. [Google Scholar] [CrossRef]
- Czifrák, K.; Lakatos, C.; Árpád Kordován, M.; Nagy, L.; Daróczi, L.; Zsuga, M.; Kéki, S. Block Copolymers of Poly(ω-Pentadecalactone) in Segmented Polyurethanes: Novel Biodegradable Shape Memory Polyurethanes. Polymers 2020, 12, 1928. [Google Scholar] [CrossRef]
- Gorbunova, M.A.; Anokhin, D.V.; Lesnichaya, V.A.; Grishchuk, A.A.; Badamshina, E.R. Optimization of Structure of Soft Block for Design of Adaptive Polyurethanes. Key Eng. Mater. 2020, 869, 273–279. [Google Scholar] [CrossRef]
- Wu, Y.H.; Wang, C.C.; Chen, C.Y. Effect of the Cyclic Structure Content on Aliphatic Polycarbonate-Based Polyurethane. Polym. J. 2021, 53, 695–702. [Google Scholar] [CrossRef]
- Mohanty, J.; Garg, H.; Gupta, P.; Alagirusamy, R.; Tripathi, B.P.; Kumar, B. Mechanically Strong and Resilient Shape Memory Polyurethane with Hexamethylene Diisocyanate as Mixing Segment. J. Intell. Mater. Syst. Struct. 2021, 32, 733–745. [Google Scholar] [CrossRef]
- Gorbunova, M.A.; Komov, E.V.; Grunin, L.Y.; Ivanova, M.S.; Abukaev, A.F.; Imamutdinova, A.M.; Ivanov, D.A.; Anokhin, D.V. The Effect of Separation of Blocks on the Crystallization Kinetics and Phase Composition of Poly(Butylene Adipate) in Multi-Block Thermoplastic Polyurethanes. Phys. Chem. Chem. Phys. 2022, 24, 902–913. [Google Scholar] [CrossRef]
- Gorbunova, M.; Komratova, V.; Grishchuk, A.; Badamshina, E. The Effect of Addition of Low-Layer Graphene Nanoparticles on Structure and Mechanical Properties of Polyurethane-Based Block Copolymers. Polym. Bull. 2019, 76, 5813–5829. [Google Scholar] [CrossRef]
- Kausar, A. Shape Memory Polyurethane/Graphene Nanocomposites: Structures, Properties, and Applications. J. Plast. Film Sheeting 2020, 36, 151–166. [Google Scholar] [CrossRef]
- Babaie, A.; Rezaei, M.; Sofla, R.L.M. Investigation of the Effects of Polycaprolactone Molecular Weight and Graphene Content on Crystallinity, Mechanical Properties and Shape Memory Behavior of Polyurethane/Graphene Nanocomposites. J. Mech. Behav. Biomed. Mater. 2019, 96, 53–68. [Google Scholar] [CrossRef]
- Auad, M.L.; Contos, V.S.; Nutt, S.; Aranguren, M.I.; Marcovich, N.E. Characterization of Nanocellulose- Reinforced Shape Memory Polyurethanes. Polym. Int. 2008, 57, 651–659. [Google Scholar] [CrossRef]
- Garces, I.T.; Aslanzadeh, S.; Boluk, Y.; Ayranci, C. Cellulose Nanocrystals (CNC) Reinforced Shape Memory Polyurethane Ribbons for Future Biomedical Applications and Design. J. Thermoplast. Compos. Mater. 2020, 33, 377–392. [Google Scholar] [CrossRef]
- Mendez, J.; Annamalai, P.K.; Eichhorn, S.J.; Rusli, R.; Rowan, S.J.; Foster, E.J.; Weder, C. Bioinspired Mechanically Adaptive Polymer Nanocomposites with Water-Activated Shape-Memory Effect. Macromolecules 2011, 44, 6827–6835. [Google Scholar] [CrossRef]
- Qi, X.; Jing, M.; Liu, Z.; Dong, P.; Liu, T.; Fu, Q. Microfibrillated Cellulose Reinforced Bio-Based Poly(Propylene Carbonate) with Dual-Responsive Shape Memory Properties. RSC Adv. 2016, 6, 7560–7567. [Google Scholar] [CrossRef]
- Korkmaz Memiş, N.; Kaplan, S. Production of Thermal and Water Responsive Shape Memory Polyurethane Nanocomposite Filaments with Cellulose Nanowhisker Incorporation. Cellulose 2021, 28, 7075–7096. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Liu, D.; Wang, W.; Liu, Y.; Zhou, S. PH-Responsive Shape Memory Poly(Ethylene Glycol)-Poly(ε-Caprolactone)-Based Polyurethane/Cellulose Nanocrystals Nanocomposite. ACS Appl. Mater. Interfaces 2015, 7, 12988–12999. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Su, Y.; Chen, B. Mechanically Adaptive and Shape-Memory Behaviour of Chitosan-Modified Cellulose Whisker/Elastomer Composites in Different PH Environments. ChemPhysChem 2014, 15, 2794–2800. [Google Scholar] [CrossRef]
- Wu, G.; Gu, Y.; Hou, X.; Li, R.; Ke, H.; Xiao, X. Hybrid Nanocomposites of Cellulose/Carbon-Nanotubes/Polyurethane with Rapidly Water Sensitive Shape Memory Effect and Strain Sensing Performance. Polymers 2019, 11, 1586. [Google Scholar] [CrossRef]
- Xu, S.; Yu, W.; Jing, M.; Huang, R.; Zhang, Q.; Fu, Q. Largely Enhanced Stretching Sensitivity of Polyurethane/Carbon Nanotube Nanocomposites via Incorporation of Cellulose Nanofiber. J. Phys. Chem. C 2017, 121, 2108–2117. [Google Scholar] [CrossRef]
- Hu, Z.; Fu, S.; Tang, A. Fabrication of Light-Triggered AuNP/CNC/SMP Nano-Composites. BioResources 2017, 12, 1982–1990. [Google Scholar] [CrossRef]
- Trache, D.; Tarchoun, A.F.; Derradji, M.; Hamidon, T.S.; Masruchin, N.; Brosse, N.; Hussin, M.H. Nanocellulose: From Fundamentals to Advanced Applications. Front. Chem. 2020, 8, 392–425. [Google Scholar] [CrossRef]
- Santos, R.F.; Ribeiro, J.C.L.; Franco de Carvalho, J.M.; Magalhães, W.L.E.; Pedroti, L.G.; Nalon, G.H.; Lima, G.E.S. de Nanofibrillated Cellulose and Its Applications in Cement-Based Composites: A Review. Constr. Build. Mater. 2021, 288, 123122–123139. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated Cellulose—Its Barrier Properties and Applications in Cellulosic Materials: A Review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef]
- Dufresne, A. Nanocellulose: A New Ageless Bionanomaterial. Mater. Today 2013, 16, 220–227. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Xie, Y.; Zhang, K. Functional Nanomaterials through Esterification of Cellulose: A Review of Chemistry and Application. Cellulose 2018, 25, 3703–3731. [Google Scholar] [CrossRef]
- Islam, M.S.; Chen, L.; Sisler, J.; Tam, K.C. Cellulose Nanocrystal (CNC)–Inorganic Hybrid Systems: Synthesis, Properties and Applications. J. Mater. Chem. B 2018, 6, 864–883. [Google Scholar] [CrossRef]
- Miyashiro, D.; Hamano, R.; Umemura, K. A Review of Applications Using Mixed Materials of Cellulose, Nanocellulose and Carbon Nanotubes. Nanomaterials 2020, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Bacakova, L.; Pajorova, J.; Tomkova, M.; Matejka, R.; Broz, A.; Stepanovska, J.; Prazak, S.; Skogberg, A.; Siljander, S.; Kallio, P. Applications of Nanocellulose/Nanocarbon Composites: Focus on Biotechnology and Medicine. Nanomaterials 2020, 10, 196. [Google Scholar] [CrossRef] [PubMed]
- Nasseri, R.; Deutschman, C.P.; Han, L.; Pope, M.A.; Tam, K.C. Cellulose Nanocrystals in Smart and Stimuli-Responsive Materials: A Review. Mater. Today Adv. 2020, 5, 100055. [Google Scholar] [CrossRef]
- Raghav, N.; Sharma, M.R.; Kennedy, J.F. Nanocellulose: A Mini-Review on Types and Use in Drug Delivery Systems. Carbohydr. Polym. Technol. Appl. 2021, 2, 100031–100041. [Google Scholar] [CrossRef]
- Abushammala, H.; Mao, J. A Review of the Surface Modification of Cellulose and Nanocellulose Using Aliphatic and Aromatic Mono- and Di-Isocyanates. Molecules 2019, 24, 2782. [Google Scholar] [CrossRef]
- Trache, D.; Hussin, M.H.; Haafiz, M.K.M.; Thakur, V.K. Recent Progress in Cellulose Nanocrystals: Sources and Production. Nanoscale 2017, 9, 1763–1786. [Google Scholar] [CrossRef]
- French, A.D. Glucose, Not Cellobiose, Is the Repeating Unit of Cellulose and Why That Is Important. Cellulose 2017, 24, 4605–4609. [Google Scholar] [CrossRef]
- Habibi, Y.; Lucia, L.A.; Rojas, O.J. Cellulose Nanocrystals: Chemistry, Self-Assembly, and Applications. Chem. Rev. 2010, 110, 3479–3500. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, V.B.; Soetaredjo, F.E.; Putro, J.N.; Santoso, S.P.; Yuliana, M.; Sunarso, J.; Ju, Y.-H.; Ismadji, S. Nanocelluloses: Sources, Pretreatment, Isolations, Modification, and Its Application as the Drug Carriers. Polymers 2021, 13, 2052. [Google Scholar] [CrossRef] [PubMed]
- Díaz, A.; Puiggalí, J. Hydrogels for Biomedical Applications: Cellulose, Chitosan, and Protein/Peptide Derivatives. Gels 2017, 3, 27. [Google Scholar] [CrossRef]
- Grunin, L.Y.; Grunin, Y.B.; Nikolskaya, E.A.; Sheveleva, N.N.; Nikolaev, I.A. An NMR Relaxation and Spin Diffusion Study of Cellulose Structure during Water Adsorption. Biophysics 2017, 62, 198–206. [Google Scholar] [CrossRef]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose Nanomaterials Review: Structure, Properties and Nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. [Google Scholar] [CrossRef]
- Grunin, Y.B.; Grunin, L.Y.; Schiraya, V.Y.; Ivanova, M.S.; Masas, D.S. Cellulose–Water System’s State Analysis by Proton Nuclear Magnetic Resonance and Sorption Measurements. Bioresour. Bioprocess. 2020, 7, 41–52. [Google Scholar] [CrossRef]
- Smirnova, L.G.; Grunin, Y.B.; Krasil’nikova, S.V.; Zaverkina, M.A.; Bakieva, D.R.; Smirnov, E.V. Study of the Structure and Sorption Properties of Some Types of Cellulose. Colloid J. 2003, 65, 778–781. [Google Scholar] [CrossRef]
- Grunin, L.Y.; Grunin, Y.B.; Talantsev, V.I.; Nikolskaya, E.A.; Masas, D.S. Features of the Structural Organization and Sorption Properties of Cellulose. Polym. Sci. Ser. A 2015, 57, 43–51. [Google Scholar] [CrossRef]
- Li, Q.; Renneckar, S. Supramolecular Structure Characterization of Molecularly Thin Cellulose i Nanoparticles. Biomacromolecules 2011, 12, 650–659. [Google Scholar] [CrossRef]
- Ding, S.Y.; Zhao, S.; Zeng, Y. Size, Shape, and Arrangement of Native Cellulose Fibrils in Maize Cell Walls. Cellulose 2014, 21, 863–871. [Google Scholar] [CrossRef]
- Barhoum, A.; Li, H.; Chen, M.; Cheng, L.; Yang, W.; Dufresne, A. Emerging Applications of Cellulose Nanofibers. In Handbook of Nanofibers; Springer International Publishing: Cham, Switzerland, 2018; pp. 1131–1157. ISBN 9783319536552. [Google Scholar]
- Khalil, H.P.S.A.; Jummaat, F.; Yahya, E.B.; Olaiya, N.G.; Adnan, A.S.; Abdat, M.; Nasir, N.A.M.; Halim, A.S.; Kumar, U.S.U.; Bairwan, R.; et al. A Review on Micro- to Nanocellulose Biopolymer Scaffold Forming for Tissue Engineering Applications. Polymers 2020, 12, 2043. [Google Scholar] [CrossRef] [PubMed]
- Delepierre, G.; Vanderfleet, O.M.; Niinivaara, E.; Zakani, B.; Cranston, E.D. Benchmarking Cellulose Nanocrystals Part II: New Industrially Produced Materials. Langmuir 2021, 37, 8393–8409. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.; Lyczko, N.; Gopakumar, D.; Maria, H.J.; Nzihou, A.; Thomas, S. Nanocellulose and Its Derivative Materials for Energy and Environmental Applications. J. Mater. Sci. 2022, 57, 6835–6880. [Google Scholar] [CrossRef]
- ISO/TC 229-TS 20477; TAPPI. Standard Terms and Their Definition for Cellulose Nanomaterial. International Organization for Standardization (ISO): Geneva, Swizerland, 2017.
- Yamashita, M.; Yoshida, M.; Matsuo, M.; Sato, S.; Yamamoto, H. Observations of Wood Cell Walls with a Scanning Probe Microscope. Mater. Sci. Appl. 2016, 7, 644–653. [Google Scholar] [CrossRef]
- Athukoralalage, S.S.; Balu, R.; Dutta, N.K.; Roy Choudhury, N. 3D Bioprinted Nanocellulose-Based Hydrogels for Tissue Engineering Applications: A Brief Review. Polymers 2019, 11, 898. [Google Scholar] [CrossRef] [PubMed]
- Phanthong, P.; Reubroycharoen, P.; Hao, X.; Xu, G.; Abudula, A.; Guan, G. Nanocellulose: Extraction and Application. Carbon Resour. Convers. 2018, 1, 32–43. [Google Scholar] [CrossRef]
- Li, N.; Lu, W.; Yu, J.; Xiao, Y.; Liu, S.; Gan, L.; Huang, J. Rod-like Cellulose Nanocrystal/Cis-Aconityl-Doxorubicin Prodrug: A Fluorescence-Visible Drug Delivery System with Enhanced Cellular Uptake and Intracellular Drug Controlled Release. Mater. Sci. Eng. C 2018, 91, 179–189. [Google Scholar] [CrossRef]
- Wang, N.; Ding, E.; Cheng, R. Thermal Degradation Behaviors of Spherical Cellulose Nanocrystals with Sulfate Groups. Polymer 2007, 48, 3486–3493. [Google Scholar] [CrossRef]
- Ait Benhamou, A.; Kassab, Z.; Nadifiyine, M.; Salim, M.H.; Sehaqui, H.; Moubarik, A.; El Achaby, M. Extraction, Characterization and Chemical Functionalization of Phosphorylated Cellulose Derivatives from Giant Reed Plant. Cellulose 2021, 28, 4625–4642. [Google Scholar] [CrossRef]
- Guimaraes, M.; Botara, V.R.; Novack, K.M.; Neto, W.P.F.; Mendes, L.M.; Tonoli, G.H.D. Preparation of Cellulose Nanofibrils from Bamboo Pulp by Mechanical Defibrillation for Their Applications in Biodegradable Composites. J. Nanosci. Nanotechnol. 2015, 15, 6751–6768. [Google Scholar] [CrossRef]
- Benhamou, K.; Kaddami, H.; Magnin, A.; Dufresne, A.; Ahmad, A. Bio-Based Polyurethane Reinforced with Cellulose Nanofibers: A Comprehensive Investigation on the Effect of Interface. Carbohydr. Polym. 2015, 122, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, N.F.; Feitosa, J.P.A.; da Gama, F.M.P.; Morais, J.P.S.; Andrade, F.K.; de Souza, M.D.S.M.; de Freitas Rosa, M. Bacterial Cellulose Nanocrystals Produced under Different Hydrolysis Conditions: Properties and Morphological Features. Carbohydr. Polym. 2017, 155, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhu, C.; Yang, J.; Nie, Y.; Chen, C.; Sun, D. Recent Advances in Bacterial Cellulose. Cellulose 2014, 21, 57–91. [Google Scholar] [CrossRef]
- Gorgieva, S.; Trček, J. Bacterial Cellulose: Production, Modification and Perspectives in Biomedical Applications. Nanomaterials 2019, 9, 1352. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Yano, H.; Nogi, M.; Eichhorn, S.J. An Estimation of the Young’s Modulus of Bacterial Cellulose Filaments. Cellulose 2008, 15, 507–513. [Google Scholar] [CrossRef]
- Meneguin, A.B.; da Silva Barud, H.; Sábio, R.M.; de Sousa, P.Z.; Manieri, K.F.; de Freitas, L.A.P.; Pacheco, G.; Alonso, J.D.; Chorilli, M. Spray-Dried Bacterial Cellulose Nanofibers: A New Generation of Pharmaceutical Excipient Intended for Intestinal Drug Delivery. Carbohydr. Polym. 2020, 249, 116838–116871. [Google Scholar] [CrossRef] [PubMed]
- Czaja, W.; Romanovicz, D.; Brown, R. malcolm Structural Investigations of Microbial Cellulose Produced in Stationary and Agitated Culture. Cellulose 2004, 11, 403–411. [Google Scholar] [CrossRef]
- Kostić, M. Development of Novel Cellulose-Based Functional Materials. Adv. Technol. 2021, 10, 73–83. [Google Scholar] [CrossRef]
- Ram, B.; Chauhan, G.S. New Spherical Nanocellulose and Thiol-Based Adsorbent for Rapid and Selective Removal of Mercuric Ions. Chem. Eng. J. 2018, 331, 587–596. [Google Scholar] [CrossRef]
- Kim, C.-W.; Kim, D.-S.; Kang, S.-Y.; Marquez, M.; Joo, Y.L. Structural Studies of Electrospun Cellulose Nanofibers. Polymer 2006, 47, 5097–5107. [Google Scholar] [CrossRef]
- Chávez-Guerrero, L.; Sepúlveda-Guzmán, S.; Silva-Mendoza, J.; Aguilar-Flores, C.; Pérez-Camacho, O. Eco-Friendly Isolation of Cellulose Nanoplatelets through Oxidation under Mild Conditions. Carbohydr. Polym. 2018, 181, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.; Pinto, F.; Lourenço, A.F.; Ferreira, P.J.T.; Louro, H.; Silva, M.J. On the Toxicity of Cellulose Nanocrystals and Nanofibrils in Animal and Cellular Models. Cellulose 2020, 27, 5509–5544. [Google Scholar] [CrossRef]
- Roman, M.; Winter, W.T. Effect of Sulfate Groups from Sulfuric Acid Hydrolysis on the Thermal Degradation Behavior of Bacterial Cellulose. Biomacromolecules 2004, 5, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.A.; Foster, E.J. Isolation of Thermally Stable Cellulose Nanocrystals from Spent Coffee Grounds via Phosphoric Acid Hydrolysis. J. Renew. Mater. 2020, 8, 187–203. [Google Scholar] [CrossRef]
- Michelin, M.; Gomes, D.G.; Romaní, A.; Polizeli, M.d.L.T.M.; Teixeira, J.A. Nanocellulose Production: Exploring the Enzymatic Route and Residues of Pulp and Paper Industry. Molecules 2020, 25, 3411. [Google Scholar] [CrossRef] [PubMed]
- Mohd Amin, K.N.; Annamalai, P.K.; Morrow, I.C.; Martin, D. Production of Cellulose Nanocrystals via a Scalable Mechanical Method. RSC Adv. 2015, 5, 57133–57140. [Google Scholar] [CrossRef]
- Saxena, I.M.; Brown, R.M. Cellulose Biosynthesis: Current Views and Evolving Concepts. Ann. Bot. 2005, 96, 9–21. [Google Scholar] [CrossRef]
- Revol, J.F.; Bradford, H.; Giasson, J.; Marchessault, R.H.; Gray, D.G. Helicoidal Self-Ordering of Cellulose Microfibrils in Aqueous Suspension. Int. J. Biol. Macromol. 1992, 14, 170–172. [Google Scholar] [CrossRef]
- Sun, Y.; Lin, L.; Pang, C.; Deng, H.; Peng, H.; Li, J.; He, B.; Liu, S. Hydrolysis of Cotton Fiber Cellulose in Formic Acid. Energy Fuels 2007, 21, 2386–2389. [Google Scholar] [CrossRef]
- Lee, S.Y.; Mohan, D.J.; Kang, I.A.; Doh, G.H.; Lee, S.; Han, S.O. Nanocellulose Reinforced PVA Composite Films: Effects of Acid Treatment and Filler Loading. Fibers Polym. 2009, 10, 77–82. [Google Scholar] [CrossRef]
- Khan, A.; Jawaid, M.; Kian, L.K.; Khan, A.A.P.; Asiri, A.M. Isolation and Production of Nanocrystalline Cellulose from Conocarpus Fiber. Polymers 2021, 13, 1835. [Google Scholar] [CrossRef] [PubMed]
- Oprea, S.; Potolinca, V.O.; Oprea, V. Physical Properties and the Ability to Disperse into Different Polar Solvents of the New Polyurethane–Cellulose Composites. J. Elastomers Plast. 2020, 52, 548–572. [Google Scholar] [CrossRef]
- Khadivi, P.; Salami-Kalajahi, M.; Roghani-Mamaqani, H.; Lotfi Mayan Sofla, R. Fabrication of Microphase-Separated Polyurethane/Cellulose Nanocrystal Nanocomposites with Irregular Mechanical and Shape Memory Properties. Appl. Phys. A 2019, 125, 779–789. [Google Scholar] [CrossRef]
- Amin, K.N.M.; Annamalai, P.K.; Martin, D. Cellulose Nanocrystals with Enhanced Thermal Stability Reinforced Thermoplastic Polyurethane. Malays. J. Anal. Sci. 2017, 21, 754–761. [Google Scholar] [CrossRef]
- Achor, M.; Oyeniyi, Y.J.; Yahaya, A. Extraction and Characterization of Microcrystalline Cellulose Obtained from the Back of the Fruit of Lageriana Siceraria (Water Gourd). J. Appl. Pharm. Sci. 2014, 4, 57–60. [Google Scholar] [CrossRef]
- Ou, H.; Chen, G.; Zhu, P.; Wei, Y.; Li, F. Preparation and Strain Sensitive Performance of Cellulose Nanofiber-Carbon Nanotubes/Thermoplastic Polyurethane Composite Films. Acta Mater. Compos. Sin. 2020, 37, 2735–2742. [Google Scholar] [CrossRef]
- Larraza, I.; Vadillo, J.; Santamaria-Echart, A.; Tejado, A.; Azpeitia, M.; Vesga, E.; Orue, A.; Saralegi, A.; Arbelaiz, A.; Eceiza, A. The Effect of the Carboxylation Degree on Cellulose Nanofibers and Waterborne Polyurethane/Cellulose Nanofiber Nanocomposites Properties. Polym. Degrad. Stab. 2020, 173, 109084–109096. [Google Scholar] [CrossRef]
- Lasseuguette, E.; Roux, D.; Nishiyama, Y. Rheological Properties of Microfibrillar Suspension of TEMPO-Oxidized Pulp. Cellulose 2008, 15, 425–433. [Google Scholar] [CrossRef]
- Barja, F. Bacterial Nanocellulose Production and Biomedical Applications. J. Biomed. Res. 2021, 35, 310–317. [Google Scholar] [CrossRef]
- Verpaalen, R.C.P.; Engels, T.; Schenning, A.P.H.J.; Debije, M.G. Stimuli-Responsive Shape Changing Commodity Polymer Composites and Bilayers. ACS Appl. Mater. Interfaces 2020, 12, 38829–38844. [Google Scholar] [CrossRef]
- Haskew, M.J.; Hardy, J.G. A Mini-Review of Shape-Memory Polymer-Based Materials: Stimuli-Responsive Shape-Memory Polymers. Johns. Matthey Technol. Rev. 2020, 64, 425–442. [Google Scholar] [CrossRef]
- Behl, M.; Razzaq, M.Y.; Lendlein, A. Multifunctional Shape-Memory Polymers. Adv. Mater. 2010, 22, 3388–3410. [Google Scholar] [CrossRef] [PubMed]
- Piegat, A.; El Fray, M. Thermoplastic Elastomers: Materials for Heart Assist Devices. Polym. Med. 2016, 46, 79–87. [Google Scholar] [CrossRef]
- Gupta, A.; Maharjan, A.; Kim, B.S. Shape Memory Polyurethane and Its Composites for Various Applications. Appl. Sci. 2019, 9, 4694. [Google Scholar] [CrossRef]
- Naureen, B.; Haseeb, A.S.M.A.; Basirun, W.J.; Muhamad, F. Recent Advances in Tissue Engineering Scaffolds Based on Polyurethane and Modified Polyurethane. Mater. Sci. Eng. C 2021, 118, 111228–111232. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, M.A.; Anokhin, D.V.; Badamshina, E.R. Recent Advances in the Synthesis and Application of Thermoplastic Semicrystalline Shape Memory Polyurethanes. Polym. Sci. Ser. B 2020, 62, 427–450. [Google Scholar] [CrossRef]
- Fisher, H.; Woolard, P.; Ross, C.; Kunkel, R.; Bohnstedt, B.N.; Liu, Y.; Lee, C.-H. Thermomechanical Data of Polyurethane Shape Memory Polymer: Considering Varying Compositions. Data Brief 2020, 32, 106294–106304. [Google Scholar] [CrossRef]
- Bratasyuk, N.A.; Zuev, V.V. The Effect Molecular Weight of Polyol Components on Shape Memory Effect of Epoxy-polyurethane Composites. Polym. Eng. Sci. 2021, 61, 2674–2690. [Google Scholar] [CrossRef]
- Kumar, B.; Noor, N.; Thakur, S.; Pan, N.; Narayana, H.; Yan, S.; Wang, F.; Shah, P. Shape Memory Polyurethane-Based Smart Polymer Substrates for Physiologically Responsive, Dynamic Pressure (Re)Distribution. ACS Omega 2019, 4, 15348–15358. [Google Scholar] [CrossRef]
- Mirtschin, N.; Pretsch, T. Designing Temperature-Memory Effects in Semicrystalline Polyurethane. RSC Adv. 2015, 5, 46307–46315. [Google Scholar] [CrossRef]
- Schönfeld, D.; Chalissery, D.; Wenz, F.; Specht, M.; Eberl, C.; Pretsch, T. Actuating Shape Memory Polymer for Thermoresponsive Soft Robotic Gripper and Programmable Materials. Molecules 2021, 26, 522. [Google Scholar] [CrossRef]
- Jin, X.; Guo, N.; You, Z.; Tan, Y. Design and Performance of Polyurethane Elastomers Composed with Different Soft Segments. Materials 2020, 13, 4991. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kang, S.-K. Principles for Controlling the Shape Recovery and Degradation Behavior of Biodegradable Shape-Memory Polymers in Biomedical Applications. Micromachines 2021, 12, 757. [Google Scholar] [CrossRef]
- Anokhin, D.V.; Gorbunova, M.A.; Abukaev, A.F.; Ivanov, D.A. Multiblock Thermoplastic Polyurethanes: In-situ Studies of Structural and Morphological Evolution under Strain. Materials 2021, 14, 3009. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.-I.; Park, H.-S.; Park, J.-H.; Knowles, J.C.; Gong, M.-S. Application of High-Strength Biodegradable Polyurethanes Containing Different Ratios of Biobased Isomannide and Poly (ϵ-Caprolactone) Diol. J. Bioact. Compat. Polym. 2013, 28, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Król, P.; Uram, Ł.; Król, B.; Pielichowska, K.; Sochacka-Piętal, M.; Walczak, M. Synthesis and Property of Polyurethane Elastomer for Biomedical Applications Based on Nonaromatic Isocyanates, Polyesters, and Ethylene Glycol. Colloid Polym. Sci. 2020, 298, 1077–1093. [Google Scholar] [CrossRef]
- Uscátegui, Y.L.; Arévalo-Alquichire, S.J.; Gómez-Tejedor, J.A.; Vallés-Lluch, A.; Díaz, L.E.; Valero, M.F. Polyurethane-Based Bioadhesive Synthesized from Polyols Derived from Castor Oil (Ricinus communis) and Low Concentration of Chitosan. J. Mater. Res. 2017, 32, 3699–3711. [Google Scholar] [CrossRef]
- Sankar, G.; Yan, N. Synthesis and Deblocking Studies of Low Temperature Heat-Curable Blocked Polymeric Methylene Diphenyl Diisocyanates. J. Macromol. Sci. Part A Pure Appl. Chem. 2015, 52, 47–55. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Yang, G.; Zheng, X.; Zhou, S. Multi-Stimulus-Responsive Shape-Memory Polymer Nanocomposite Network Cross-Linked by Cellulose Nanocrystals. ACS Appl. Mater. Interfaces 2015, 7, 4118–4126. [Google Scholar] [CrossRef]
- Raschip, I.E.; Moldovan, L.; Stefan, L.; Oancea, A.; Vasile, C. Compatibility and Cytotoxicity Testing of Some New Biomaterials Based on Polyurethane and Hydroxypropylcellulose Blends. Optoelectron. Adv. Mater.-Rapid Commun. 2009, 3, 1336–1342. [Google Scholar]
- Karpov, S.V.; Dzhalmukhanova, A.S.; Chernyayev, D.A.; Lodygina, V.P.; Firsova, A.I.; Badamshina, E.R. The Investigation of Triethylammonium Carboxylates Influence on the Kinetics of Urethane Formation Processing during Waterborne Polyurethane Synthesis. Polym. Adv. Technol. 2021, 32, 2727–2734. [Google Scholar] [CrossRef]
- Karpov, S.V.; Lodygina, V.P.; Komratova, V.V.; Dzhalmukhanova, A.S.; Malkov, G.V.; Badamshina, E.R. Kinetics of Urethane Formation from Isophorone Diisocyanate: The Catalyst and Solvent Effects. Kinet. Catal. 2016, 57, 422–428. [Google Scholar] [CrossRef]
- Singh, A.; Kumar, R.; Soni, P.K.; Singh, V. Investigation of the Effect of Diisocyanate on the Thermal Degradation Behavior and Degradation Kinetics of Polyether-Based Polyurethanes. J. Macromol. Sci. Part B Phys. 2020, 59, 775–795. [Google Scholar] [CrossRef]
- Javaid, M.A.; Zia, K.M.; Khera, R.A.; Jabeen, S.; Mumtaz, I.; Younis, M.A.; Shoaib, M.; Bhatti, I.A. Evaluation of Cytotoxicity, Hemocompatibility and Spectral Studies of Chitosan Assisted Polyurethanes Prepared with Various Diisocyanates. Int. J. Biol. Macromol. 2019, 129, 116–126. [Google Scholar] [CrossRef]
- Uscátegui, Y.L.; Díaz, L.E.; Valero, M.F. In Vitro and in Vivo Biocompatibility of Polyurethanes Synthesized with Castor Oil Polyols for Biomedical Devices. J. Mater. Res. 2019, 34, 519–531. [Google Scholar] [CrossRef]
- Krol, P. Synthesis Methods, Chemical Structures and Phase Structures of Linear Polyurethanes. Properties and Applications of Linear Polyurethanes in Polyurethane Elastomers, Copolymers and Ionomers. Prog. Mater. Sci. 2007, 52, 915–1015. [Google Scholar] [CrossRef]
- Anokhin, D.V.; Gorbunova, M.A.; Estrin, Y.I.; Komratova, V.V.; Badamshina, E.R. The Role of Fast and Slow Processes in the Formation of Structure and Properties of Thermoplastic Polyurethanes. Phys. Chem. Chem. Phys. 2016, 18, 31769–31776. [Google Scholar] [CrossRef]
- Gorbunova, M.A.; Shukhardin, D.M.; Lesnichaya, V.A.; Badamshina, E.R.; Anokhin, D.V. New Polyurethane Urea Thermoplastic Elastomers with Controlled Mechanical and Thermal Properties for Medical Applications. Key Eng. Mater. 2019, 816, 187–191. [Google Scholar] [CrossRef]
- Li, L.; Xu, L.; Ding, W.; Lu, H.; Zhang, C.; Liu, T. Molecular-Engineered Hybrid Carbon Nanofillers for Thermoplastic Polyurethane Nanocomposites with High Mechanical Strength and Toughness. Compos. Part B Eng. 2019, 177, 107381–107391. [Google Scholar] [CrossRef]
- Yao, Z.; Wu, D.; Chen, C.; Zhang, M. Creep Behavior of Polyurethane Nanocomposites with Carbon Nanotubes. Compos. Part A Appl. Sci. Manuf. 2013, 50, 65–72. [Google Scholar] [CrossRef]
- Marcovich, N.E.; Auad, M.L.; Bellesi, N.E.; Nutt, S.R.; Aranguren, M.I. Cellulose Micro/Nanocrystals Reinforced Polyurethane. J. Mater. Res. 2006, 21, 870–881. [Google Scholar] [CrossRef]
- Bras, J.; Viet, D.; Bruzzese, C.; Dufresne, A. Correlation between Stiffness of Sheets Prepared from Cellulose Whiskers and Nanoparticles Dimensions. Carbohydr. Polym. 2011, 84, 211–215. [Google Scholar] [CrossRef]
- Iwamoto, S.; Nakagaito, A.N.; Yano, H. Nano-Fibrillation of Pulp Fibers for the Processing of Transparent Nanocomposites. Appl. Phys. A 2007, 89, 461–466. [Google Scholar] [CrossRef]
- Nogi, M.; Iwamoto, S.; Nakagaito, A.N.; Yano, H. Optically Transparent Nanofiber Paper. Adv. Mater. 2009, 21, 1595–1598. [Google Scholar] [CrossRef]
- Pei, A.; Malho, J.M.; Ruokolainen, J.; Zhou, Q.; Berglund, L.A. Strong Nanocomposite Reinforcement Effects in Polyurethane Elastomer with Low Volume Fraction of Cellulose Nanocrystals. Macromolecules 2011, 44, 4422–4427. [Google Scholar] [CrossRef]
- Prataviera, R.; Pollet, E.; Bretas, R.E.S.; Avérous, L.; de Almeida Lucas, A. Melt Processing of Nanocomposites of Cellulose Nanocrystals with Biobased Thermoplastic Polyurethane. J. Appl. Polym. Sci. 2021, 138, 12–20. [Google Scholar] [CrossRef]
- Noormohammadi, F.; Nourany, M.; Mir Mohamad Sadeghi, G.; Wang, P.-Y.; Shahsavarani, H. The Role of Cellulose Nanowhiskers in Controlling Phase Segregation, Crystallization and Thermal Stimuli Responsiveness in PCL-PEGx-PCL Block Copolymer-Based PU for Human Tissue Engineering Applications. Carbohydr. Polym. 2021, 252, 117219–117227. [Google Scholar] [CrossRef]
- Du, W.; Zhang, Z.; Yin, C.; Ge, X.; Shi, L. Preparation of Shape Memory Polyurethane/Modified Cellulose Nanocrystals Composites with Balanced Comprehensive Performances. Polym. Adv. Technol. 2021, 32, 4710–4720. [Google Scholar] [CrossRef]
- Bi, H.; Ren, Z.; Ye, G.; Sun, H.; Guo, R.; Jia, X.; Xu, M. Fabrication of Cellulose Nanocrystal Reinforced Thermoplastic Polyurethane/Polycaprolactone Blends for Three-Dimension Printing Self-Healing Nanocomposites. Cellulose 2020, 27, 8011–8026. [Google Scholar] [CrossRef]
- Nicharat, A.; Shirole, A.; Foster, E.J.; Weder, C. Thermally Activated Shape Memory Behavior of Melt-Mixed Polyurethane/Cellulose Nanocrystal Composites. J. Appl. Polym. Sci. 2017, 134, 45033–45043. [Google Scholar] [CrossRef]
- Mabrouk, A.B.; Kaddami, H.; Boufi, S.; Erchiqui, F.; Dufresne, A. Cellulosic Nanoparticles from Alfa Fibers (Stipa Tenacissima): Extraction Procedures and Reinforcement Potential in Polymer Nanocomposites. Cellulose 2012, 19, 843–853. [Google Scholar] [CrossRef]
- Bendahou, A.; Kaddami, H.; Dufresne, A. Investigation on the Effect of Cellulosic Nanoparticles’ Morphology on the Properties of Natural Rubber Based Nanocomposites. Eur. Polym. J. 2010, 46, 609–620. [Google Scholar] [CrossRef]
- Pandey, J.K.; Lee, C.S.; Ahn, S.-H. Preparation and Properties of Bio-Nanoreinforced Composites from Biodegradable Polymer Matrix and Cellulose Whiskers. J. Appl. Polym. Sci. 2010, 115, 2493–2501. [Google Scholar] [CrossRef]
- Eyley, S.; Thielemans, W. Surface Modification of Cellulose Nanocrystals. Nanoscale 2014, 6, 7764–7779. [Google Scholar] [CrossRef]
- Shi, Z.; Li, S.; Li, M.; Gan, L.; Huang, J. Surface Modification of Cellulose Nanocrystals towards New Materials Development. J. Appl. Polym. Sci. 2021, 138, e51555–e51576. [Google Scholar] [CrossRef]
- Shirole, A.; Nicharat, A.; Perotto, C.U.; Weder, C. Tailoring the Properties of a Shape-Memory Polyurethane via Nanocomposite Formation and Nucleation. Macromolecules 2018, 51, 1841–1849. [Google Scholar] [CrossRef]
- Cai, C.; Wei, Z.; Wang, X.; Mei, C.; Fu, Y.; Zhong, W.H. Novel Double-Networked Polyurethane Composites with Multi-Stimuli Responsive Functionalities. J. Mater. Chem. A 2018, 6, 17457–17472. [Google Scholar] [CrossRef]
- Rueda, L.; Fernández d’Arlas, B.; Zhou, Q.; Berglund, L.A.; Corcuera, M.A.; Mondragon, I.; Eceiza, A. Isocyanate-Rich Cellulose Nanocrystals and Their Selective Insertion in Elastomeric Polyurethane. Compos. Sci. Technol. 2011, 71, 1953–1960. [Google Scholar] [CrossRef]
- Tian, D.; Wang, F.; Yang, Z.; Niu, X.; Wu, Q.; Sun, P. High-Performance Polyurethane Nanocomposites Based on UPy-Modified Cellulose Nanocrystals. Carbohydr. Polym. 2019, 219, 191–200. [Google Scholar] [CrossRef]
- Girouard, N.M.; Xu, S.; Schueneman, G.T.; Shofner, M.L.; Meredith, J.C. Site-Selective Modification of Cellulose Nanocrystals with Isophorone Diisocyanate and Formation of Polyurethane-CNC Composites. ACS Appl. Mater. Interfaces 2016, 8, 1458–1467. [Google Scholar] [CrossRef]
- Abushammala, H.; Mao, J. Impact of the Surface Properties of Cellulose Nanocrystals on the Crystallization Kinetics of Poly(Butylene Succinate). Crystals 2020, 10, 196. [Google Scholar] [CrossRef]
- Abushammala, H. Nano-Brushes of Alcohols Grafted onto Cellulose Nanocrystals for Reinforcing Poly(Butylene Succinate): Impact of Alcohol Chain Length on Interfacial Adhesion. Polymers 2020, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Abushammala, H. On the Para/Ortho Reactivity of Isocyanate Groups during the Carbamation of Cellulose Nanocrystals Using 2,4-Toluene Diisocyanate. Polymers 2019, 11, 1164. [Google Scholar] [CrossRef]
- Abushammala, H. A Simple Method for the Quantification of Free Isocyanates on the Surface of Cellulose Nanocrystals upon Carbamation Using Toluene Diisocyanate. Surfaces 2019, 2, 444–454. [Google Scholar] [CrossRef]
- Gwon, J.G.; Cho, H.J.; Chun, S.J.; Lee, S.; Wu, Q.; Li, M.C.; Lee, S.Y. Mechanical and Thermal Properties of Toluene Diisocyanate-Modified Cellulose Nanocrystal Nanocomposites Using Semi-Crystalline Poly(Lactic Acid) as a Base Matrix. RSC Adv. 2016, 6, 73879–73886. [Google Scholar] [CrossRef]
- Semsarzadeh, M.A.; Navarchian, A.H. Kinetic Study of the Bulk Reaction between Tdi and Ppg in Presence of Dbtdl and Feaa Catalysts Using Quantitative Ftir Spectroscopy. J. Polym. Eng. 2003, 23, 225–240. [Google Scholar] [CrossRef]
- Zaverkina, M.A.; Lodygina, V.P.; Komratova, V.V.; Stovbun, E.V.; Badamshina, E.R. Kinetics of Diisocyanate Reactions with Chain-Extending Agents. Polym. Sci. Ser. A 2006, 48, 382–387. [Google Scholar] [CrossRef]
- Špirková, M.; Kubín, M.; Dušek, K. Side Reactions in the Formation of Polyurethanes: Model Reactions Between Phenylisocyanate and 1-Butanol. J. Macromol. Sci. Part A-Chem. 1987, 24, 1151–1166. [Google Scholar] [CrossRef]
- Morandi, G.; Thielemans, W. Synthesis of Cellulose Nanocrystals Bearing Photocleavable Grafts by ATRP. Polym. Chem. 2012, 3, 1402–1407. [Google Scholar] [CrossRef]
- Karpov, S.V.; Lodygina, V.P.; Komratova, V.V.; Dzhalmukhanova, A.S.; Malkov, G.V.; Badamshina, E.R. Kinetics of Urethane Formation from Isophorone Diisocyanate: The Alcohol Nature Effect. Kinet. Catal. 2016, 57, 319–325. [Google Scholar] [CrossRef]
- Lomölder, R.; Plogmann, F.; Speier, P. Selectivity of Isophorone Diisocyanate in the Urethane Reaction Influence of Temperature, Catalysis, and Reaction Partners. J. Coat. Technol. 1997, 69, 51–57. [Google Scholar] [CrossRef]
- De Mesquita, J.P.; Donnici, C.L.; Pereira, F.V. Biobased Nanocomposites from Layer-by-Layer Assembly of Cellulose Nanowhiskers with Chitosan. Biomacromolecules 2010, 11, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Long, L.-Y.; Weng, Y.-X.; Wang, Y.-Z. Cellulose Aerogels: Synthesis, Applications, and Prospects. Polymers 2018, 10, 623. [Google Scholar] [CrossRef] [PubMed]
- Capadona, J.R.; Van Den Berg, O.; Capadona, L.A.; Schroeter, M.; Rowan, S.J.; Tyler, D.J.; Weder, C. A Versatile Approach for the Processing of Polymer Nanocomposites with Self-Assembled Nanofibre Templates. Nat. Nanotechnol. 2007, 2, 765–769. [Google Scholar] [CrossRef]
- Shi, Z.; Huang, J.; Liu, C.; Ding, B.; Kuga, S.; Cai, J.; Zhang, L. Three-Dimensional Nanoporous Cellulose Gels as a Flexible Reinforcement Matrix for Polymer Nanocomposites. ACS Appl. Mater. Int. 2015, 7, 22990–22998. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wei, P.; Huang, J.; Xu, D.; Zhong, Y.; Hu, L.; Zhang, L.; Cai, J. Mechanically Strong Shape-Memory and Solvent-Resistant Double-Network Polyurethane/Nanoporous Cellulose Gel Nanocomposites. ACS Sustain. Chem. Eng. 2019, 7, 15974–15982. [Google Scholar] [CrossRef]
- Amin, K.N.M.; Amiralian, N.; Annamalai, P.K.; Edwards, G.; Chaleat, C.; Martin, D.J. Scalable Processing of Thermoplastic Polyurethane Nanocomposites Toughened with Nanocellulose. Chem. Eng. J. 2016, 302, 406–416. [Google Scholar] [CrossRef]
- Seydibeyoǧlu, M.Ö.; Misra, M.; Mohanty, A.; Blaker, J.J.; Lee, K.Y.; Bismarck, A.; Kazemizadeh, M. Green Polyurethane Nanocomposites from Soy Polyol and Bacterial Cellulose. J. Mater. Sci. 2013, 48, 2167–2175. [Google Scholar] [CrossRef]
- Özgür Seydibeyoǧlu, M.; Oksman, K. Novel Nanocomposites Based on Polyurethane and Micro Fibrillated Cellulose. Compos. Sci. Technol. 2008, 68, 908–914. [Google Scholar] [CrossRef]
- Khadivi, P.; Salami-Kalajahi, M.; Roghani-Mamaqani, H.; Sofla, R.L.M. Polydimethylsiloxane-based Polyurethane/Cellulose Nanocrystal Nanocomposites: From Structural Properties Toward Cytotoxicity. Silicon 2021, 14, 1695–1703. [Google Scholar] [CrossRef]
- Saralegi, A.; Gonzalez, M.L.; Valea, A.; Eceiza, A.; Corcuera, M.A. The Role of Cellulose Nanocrystals in the Improvement of the Shape-Memory Properties of Castor Oil-Based Segmented Thermoplastic Polyurethanes. Compos. Sci. Technol. 2014, 92, 27–33. [Google Scholar] [CrossRef]
- Lee, M.; Heo, M.H.; Lee, H.H.; Kim, Y.W.; Shin, J. Tunable Softening and Toughening of Individualized Cellulose Nanofibers-Polyurethane Urea Elastomer Composites. Carbohydr. Polym. 2017, 159, 125–135. [Google Scholar] [CrossRef]
- Rueda, L.; Saralegi, A.; Fernández-d’Arlas, B.; Zhou, Q.; Alonso-Varona, A.; Berglund, L.A.; Mondragon, I.; Corcuera, M.A.; Eceiza, A. In-situ Polymerization and Characterization of Elastomeric Polyurethane-Cellulose Nanocrystal Nanocomposites. Cell Response Evaluation. Cellulose 2013, 20, 1819–1828. [Google Scholar] [CrossRef]
- Rueda, L.; Saralegui, A.; Fernández D’Arlas, B.; Zhou, Q.; Berglund, L.A.; Corcuera, M.A.; Mondragon, I.; Eceiza, A. Cellulose Nanocrystals/Polyurethane Nanocomposites. Study from the Viewpoint of Microphase Separated Structure. Carbohydr. Polym. 2013, 92, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Auad, M.L.; Mosiewicki, M.A.; Richardson, T.; Aranguren, M.I.; Marcovich, N.E. Nanocomposites Made from Cellulose Nanocrystals and Tailored Segmented Polyurethanes. J. Appl. Polym. Sci. 2010, 115, 1215–1225. [Google Scholar] [CrossRef]
- Shahrousvand, E.; Shahrousvand, M. Preparation of Polyurethane/Poly (2-Hydroxyethyl Methacrylate) Semi-IPNs Containing Cellulose Nanocrystals for Biomedical Applications. Mater. Today Commun. 2021, 27, 102421–102432. [Google Scholar] [CrossRef]
- Shahrousvand, M.; Ghollasi, M.; Zarchi, A.A.K.; Salimi, A. Osteogenic Differentiation of HMSCs on Semi-Interpenetrating Polymer Networks of Polyurethane/Poly(2-hydroxyethyl Methacrylate)/Cellulose Nanowhisker Scaffolds. Int. J. Biol. Macromol. 2019, 138, 262–271. [Google Scholar] [CrossRef]
- Saralegi, A.; Rueda, L.; Martin, L.; Arbelaiz, A.; Eceiza, A.; Corcuera, M.A. From Elastomeric to Rigid Polyurethane/Cellulose Nanocrystal Bionanocomposites. Compos. Sci. Technol. 2013, 88, 39–47. [Google Scholar] [CrossRef]
- Brakat, A.; Zhu, H. Nanocellulose-Graphene Hybrids: Advanced Functional Materials as Multifunctional Sensing Platform. Nano-Micro Lett. 2021, 13, 94–131. [Google Scholar] [CrossRef]
- Liu, H.; Song, J.; Shang, S.; Song, Z.; Wang, D. Cellulose Nanocrystal/Silver Nanoparticle Composites as Bifunctional Nanofillers within Waterborne Polyurethane. ACS Appl. Mater. Interfaces 2012, 4, 2413–2419. [Google Scholar] [CrossRef]
- Wong, B.S.; Yoong, S.L.; Jagusiak, A.; Panczyk, T.; Ho, H.K.; Ang, W.H.; Pastorin, G. Carbon Nanotubes for Delivery of Small Molecule Drugs. Adv. Drug Deliv. Rev. 2013, 65, 1964–2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Anoshkin, I.V.; Nasibulin, A.G.; Korhonen, J.T.; Seitsonen, J.; Pere, J.; Kauppinen, E.I.; Ras, R.H.A.; Ikkala, O. Modifying Native Nanocellulose Aerogels with Carbon Nanotubes for Mechanoresponsive Conductivity and Pressure Sensing. Adv. Mater. 2013, 25, 2428–2432. [Google Scholar] [CrossRef] [PubMed]
- Olivier, C.; Moreau, C.; Bertoncini, P.; Bizot, H.; Chauvet, O.; Cathala, B. Cellulose Nanocrystal-Assisted Dispersion of Luminescent Single-Walled Carbon Nanotubes for Layer-by-Layer Assembled Hybrid Thin Films. Langmuir 2012, 28, 12463–12471. [Google Scholar] [CrossRef] [PubMed]
- Hamedi, M.M.; Hajian, A.; Fall, A.B.; Hkansson, K.; Salajkova, M.; Lundell, F.; Wgberg, L.; Berglund, L.A. Highly Conducting, Strong Nanocomposites Based on Nanocellulose-Assisted Aqueous Dispersions of Single-Wall Carbon Nanotubes. ACS Nano 2014, 8, 2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.-S.; Zeng, H.-X.; Wu, J.; Dong, L.-Y.; Zhu, J.-T.; Xue, Z.-G.; Zhou, X.-P.; Xie, X.-L.; Mai, Y.-W. Biocompatible Reduced Graphene Oxide Sheets with Superior Water Dispersibility Stabilized by Cellulose Nanocrystals and Their Polyethylene Oxide Composites. Green Chem. 2016, 18, 1674–1683. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, H.; He, X.; Hu, Y.; Zhu, E.; Gao, Y.; Liu, D.; Shi, Z.; Li, J.; Yang, Q.; et al. Flexible Electronic Skin Sensor Based on Regenerated Cellulose/Carbon Nanotube Composite Films. Cellulose 2020, 27, 10199–10211. [Google Scholar] [CrossRef]
- Bai, Y.; Jiang, C.; Wang, Q.; Wang, T. Multi-Shape-Memory Property Study of Novel Poly(ε-Caprolactone)/Ethyl Cellulose Polymer Networks. Macromol. Chem. Phys. 2013, 214, 2465–2472. [Google Scholar] [CrossRef]
- Wang, W.; Liu, D.; Lu, L.; Chen, H.; Gong, T.; Lv, J.; Zhou, S. The Improvement of the Shape Memory Function of Poly(ϵ-Caprolactone)/Nano-Crystalline Cellulose Nanocomposites via Recrystallization under a High-Pressure Environment. J. Mater. Chem. A 2016, 4, 5984–5992. [Google Scholar] [CrossRef]
- Du, Y.; Li, D.; Liu, L.; Gai, G. Recent Achievements of Self-Healing Graphene/Polymer Composites. Polymers 2018, 10, 114. [Google Scholar] [CrossRef]
- Wu, T.; Frydrych, M.; O’Kelly, K.; Chen, B. Poly(Glycerol Sebacate Urethane)–Cellulose Nanocomposites with Water-Active Shape-Memory Effects. Biomacromolecules 2014, 15, 2663–2671. [Google Scholar] [CrossRef]
- Capadona, J.R.; Shanmuganathan, K.; Tyler, D.J.; Rowan, S.J.; Weder, C. Stimuli-Responsive Polymer Nanocomposites Inspired by the Sea Cucumber Dermis. Science 2008, 319, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yang, G.; Jing, M.; Fu, Q.; Chiu, F.C. Microfibrillated Cellulose-Reinforced Bio-Based Poly(Propylene Carbonate) with Dual Shape Memory and Self-Healing Properties. J. Mater. Chem. A 2014, 2, 20393–20401. [Google Scholar] [CrossRef]
- Tan, L.; Hu, J.; Ying Rena, K.; Zhu, Y.; Liu, P. Quick Water-Responsive Shape Memory Hybrids with Cellulose Nanofibers. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 767–775. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, J.; Luo, H.; Young, R.J.; Deng, L.; Zhang, S.; Fan, Y.; Ye, G. Rapidly Switchable Water-Sensitive Shape-Memory Cellulose/Elastomer Nano-Composites. Soft Matter 2012, 8, 2509. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, Z.; Liu, Z.; Kang, H.; Liu, Y. Cellulose Nanofibers/Polyurethane Shape Memory Composites with Fast Water-Responsivity. J. Mater. Chem. B 2018, 6, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Hu, J.; Zhu, Y. Polymeric Shape Memory Nanocomposites with Heterogeneous Twin Switches. Macromol. Chem. Phys. 2011, 212, 1981–1986. [Google Scholar] [CrossRef]
- Cao, X.; Xu, C.; Wang, Y.; Liu, Y.; Liu, Y.; Chen, Y. New Nanocomposite Materials Reinforced with Cellulose Nanocrystals in Nitrile Rubber. Polym. Test. 2013, 32, 819–826. [Google Scholar] [CrossRef]
- Ghosal, K.; Agatemor, C.; Tucker, N.; Kny, E.; Thomas, S. Chapter 1. Electrical Spinning to Electrospinning: A Brief History. In Electrospinning: From Basic Research to Commercialization; The Royal Society of Chemistry: London, UK, 2018; pp. 1–23. ISBN 978-1-78801-100-6. [Google Scholar]
- Fleige, E.; Quadir, M.A.; Haag, R. Stimuli-Responsive Polymeric Nanocarriers for the Controlled Transport of Active Compounds: Concepts and Applications. Adv. Drug Deliv. Rev. 2012, 64, 866–884. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A. Chitins and Chitosans for the Repair of Wounded Skin, Nerve, Cartilage and Bone. Carbohydr. Polym. 2009, 76, 167–182. [Google Scholar] [CrossRef]
- Araki, J.; Wada, M.; Kuga, S.; Okano, T. Flow Properties of Microcrystalline Cellulose Suspension Prepared by Acid Treatment of Native Cellulose. Colloids Surf. A Physicochem. Eng. Asp. 1998, 142, 75–82. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Liu, Y.; Gong, T.; Wang, L.; Zhou, S. Highly PH-Sensitive Polyurethane Exhibiting Shape Memory and Drug Release. Polym. Chem. 2014, 5, 5168–5174. [Google Scholar] [CrossRef]
- Way, A.E.; Hsu, L.; Shanmuganathan, K.; Weder, C.; Rowan, S.J. PH-Responsive Cellulose Nanocrystal Gels and Nanocomposites. ACS Macro Lett. 2012, 1, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz Memiş, N.; Kaplan, S. Dual Responsive Wool Fabric by Cellulose Nanowhisker Reinforced Shape Memory Polyurethane. J. Appl. Polym. Sci. 2020, 137, 28–38. [Google Scholar] [CrossRef]
- Sukhkhawuttigit, S.; Ummartyotin, S.; Infahsaeng, Y. Development of Chemically Grafted Multiwall Carbon Nanotube onto Cellulose Fiber Sheet and Polyurethane Based Resin Composite for an Active Paper. J. Met. Mater. Miner. 2021, 31, 110–117. [Google Scholar] [CrossRef]
- Shrestha, S.; Shrestha, B.K.; Lee, J.; Joong, O.K.; Kim, B.S.; Park, C.H.; Kim, C.S. A Conducting Neural Interface of Polyurethane/Silk-Functionalized Multiwall Carbon Nanotubes with Enhanced Mechanical Strength for Neuroregeneration. Mater. Sci. Eng. C 2019, 102, 511–523. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terminology and Nomenclature of Cellulose Nanomaterials | Diameter (nm) | Length (nm) | Aspect Ratio (Length/Diameter) |
|---|---|---|---|
| Cellulose NanoCrystal (CNC) | 3–10 | <500 | ˃5 |
| Cellulose NanoFibril (CNF) | 5–30 | 100–600 | ˃50 |
| Bacterial NanoCellulose (BNC) | 10–40 | ˃1000 | 100–150 |
| Cellulose MicroCrystal (CMC) | 10,000–15,000 | ˃1000 | <2 |
| Cellulose MicroFibril (CMF) | 10–100 | 500–10,000 | 50–500 |
| Amorphous NanoCellulose (ANC) | 80–120 | - | - |
| Cellulose NanoYarn (CNY) | 100–1000 | several microns | - |
| Amorphous cellulose NanoParticles (ANP) | 3 | - | - |
| Composite and Method of Production | Composition of Thermoplastic Polyurethanes | Nanoparticle Size and Concentration | Film Thickness (mm) | Response Temperature (°C) | Response Time (min) | Reference |
|---|---|---|---|---|---|---|
| Texin 985/CNF mixing in solution | PTMG; MDI; 1,4-butane diol | datdiameter 15 nm, length 250 nm, aspect ratio 16.67 30 wt% | 0.1–0.2 | room | 1 | [187] |
| TPU/CNC mixing in solution | PCL (Mn ~4000 Da); MDI; 1,4-butane diol | - | - | 65 | 2–3 | [188] |
| TPU HT751/CNC mixing in solution | polymethyl methacrylate | diameter 18.5 ± 5.9 nm, length 272 ± 87 nm, aspect ratio 15, 4.6 wt% | 0.2 | room | 10 | [186] |
| TPU HT-751/CNC electrospinning | polymethyl methacrylate | diameter 50–650 nm, 24.0 wt% width 200–300 nm | - | room | 2 | [185] |
| TPU-CNC In situ | HMDI; glycerol, sebacic acid | length <200 nm, aspect ratio 11.8, 18.6 vol% width 18 nm, | 0.15–0.25 | 37 (pH = 7.4), 22 (water) | 30 | [182] |
| Texin 985-CNC In situ | PTMG (Mn ~2000 Da), MDI; 1,4-butane diol | length 200 nm, aspect ratio ~11, 20 wt% | 0.2–0.3 | 37 | 24 h | [20] |
| TPU-CNF In situ | PEG (Mw ~2000 Da); TDI; 3,3-dichloro-4,4-diamino-diphenylmethanelithium | diameter 20 nm 9 wt% | - | 37 | 1 | [158] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorbunova, M.; Grunin, L.; Morris, R.H.; Imamutdinova, A. Nanocellulose-Based Thermoplastic Polyurethane Biocomposites with Shape Memory Effect. J. Compos. Sci. 2023, 7, 168. https://doi.org/10.3390/jcs7040168
Gorbunova M, Grunin L, Morris RH, Imamutdinova A. Nanocellulose-Based Thermoplastic Polyurethane Biocomposites with Shape Memory Effect. Journal of Composites Science. 2023; 7(4):168. https://doi.org/10.3390/jcs7040168
Chicago/Turabian StyleGorbunova, Marina, Leonid Grunin, Robert H. Morris, and Arina Imamutdinova. 2023. "Nanocellulose-Based Thermoplastic Polyurethane Biocomposites with Shape Memory Effect" Journal of Composites Science 7, no. 4: 168. https://doi.org/10.3390/jcs7040168
APA StyleGorbunova, M., Grunin, L., Morris, R. H., & Imamutdinova, A. (2023). Nanocellulose-Based Thermoplastic Polyurethane Biocomposites with Shape Memory Effect. Journal of Composites Science, 7(4), 168. https://doi.org/10.3390/jcs7040168