A Parallel Software Pipeline for DMET Microarray Genotyping Data Analysis




Abstract
:1. Introduction
2. Related Work
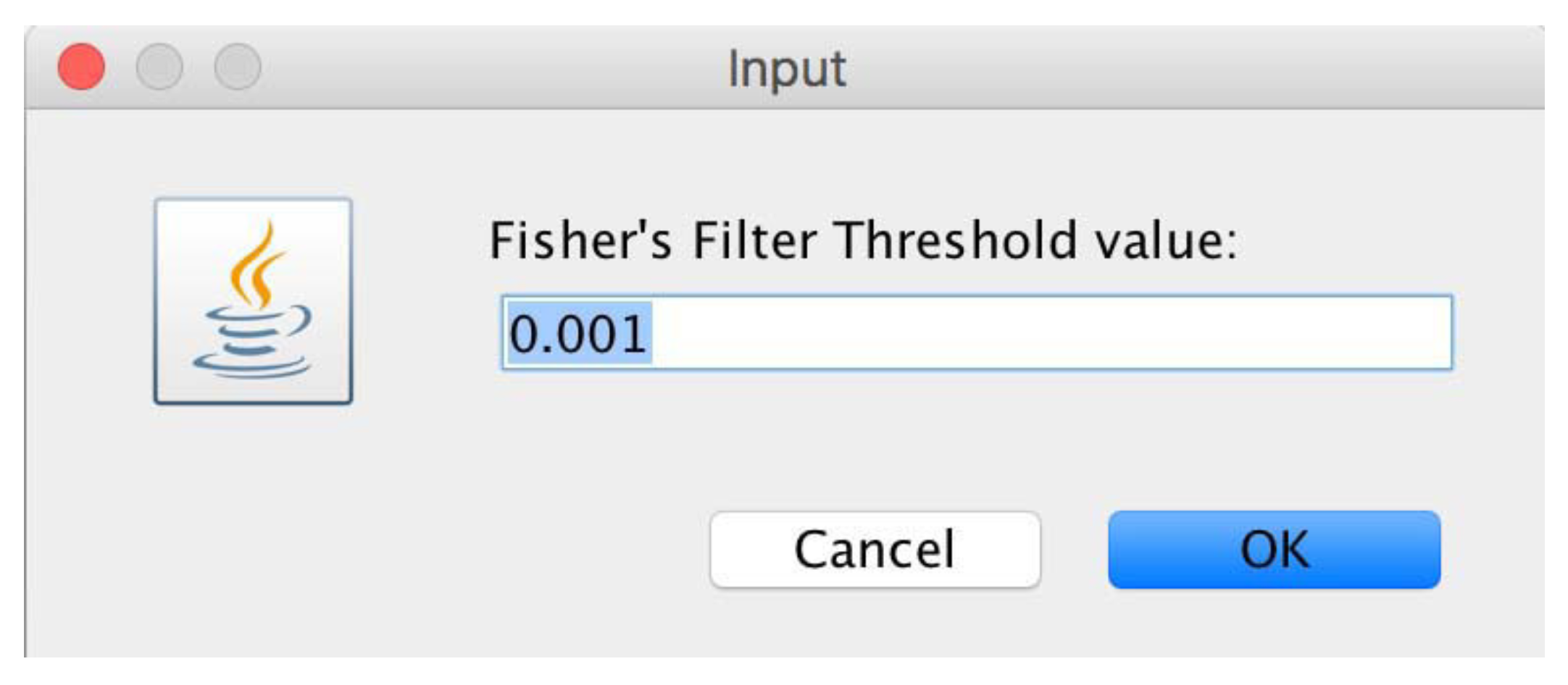
- DMET-Analyzer [8] is a tool for the automatic association analysis of the difference between the genomics characteristics and the clinical status of patients, e.g., the different response to drugs. DMET-Analyzer can investigate the association between the presence or absence of SNP related to the groups of patients (e.g., RESPONDER or NOT-RESPONDER) by using the well known Fisher’s test. To improve the statistical significance of results DMET-Analyzer implements the Bonferroni and False Discovery Rate (FDR) statistical correctors. Finally, DMET-Analyzer can retrieve information provided by Affymetrix libraries and with links to the dbSNP database and the PharmGKB pharmacogenomics database to annotates the computed relevant SNPs.
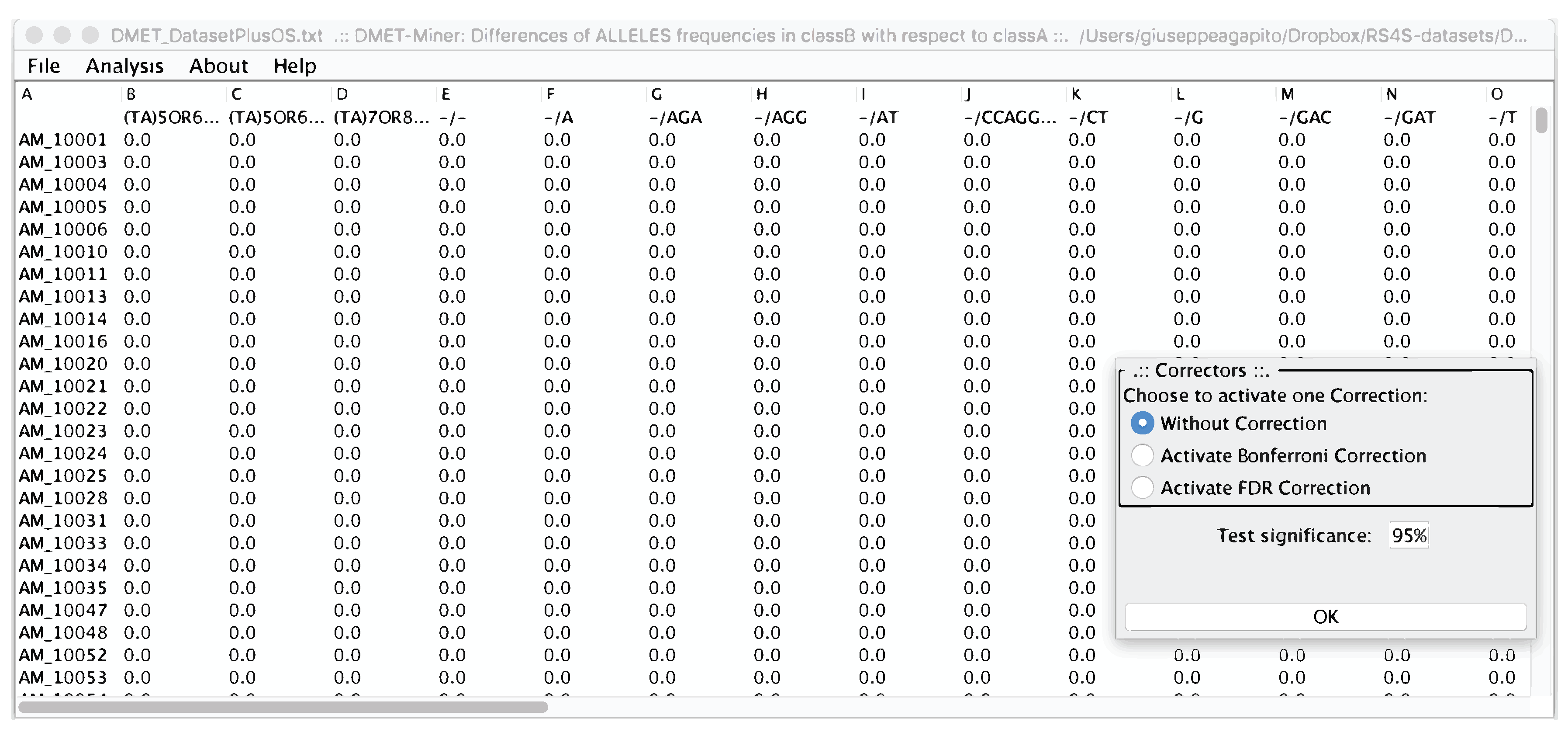
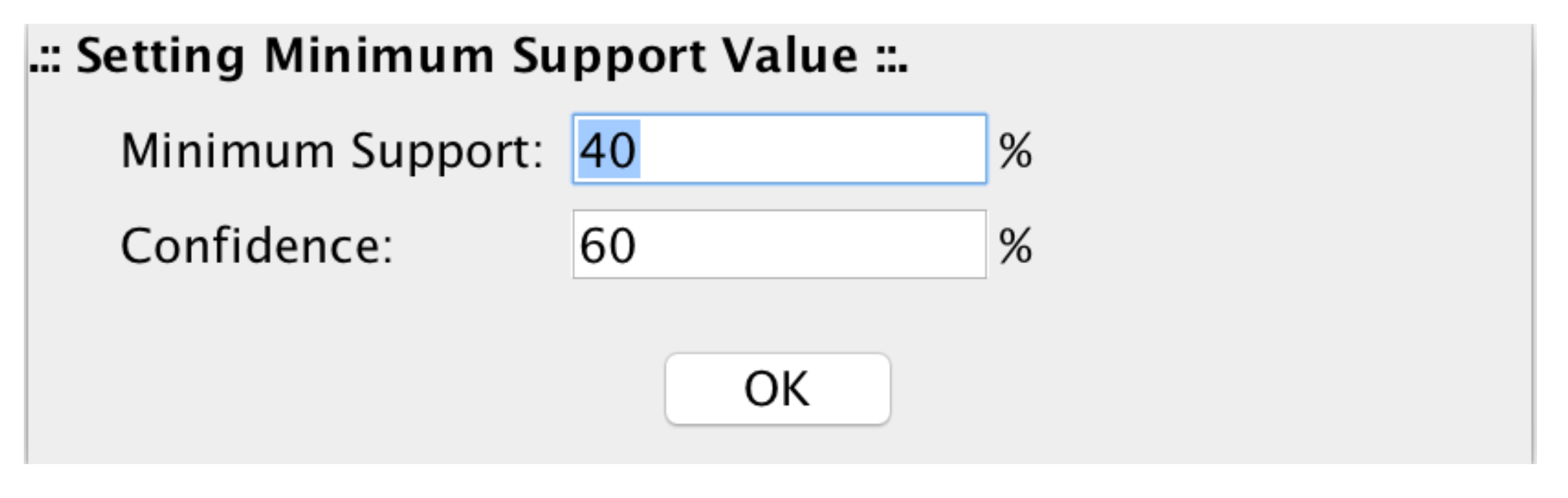
- DMET-Miner [9] is a tool developed to automatically and easily mine association rules from DMET datasets that can highlight the presence–absence of multiple SNPs related to the clinical condition of a subject. For example, the combination of SNPs responsible for the different response to drugs by an individual.
- OS-Analyzer [10] is a software tool to analyze DMET datasets annotated with clinical data such as OS and PFS. The primary function of OSA is the automatic computation and visualization of OS and PFS curves due to the presence of specific ADME genes, from a whole DMET dataset. Results are conveyed to the user ranked by statistical relevance obtained computed the area under the curves by using log-rank test.
- coreSNP [11] is the parallel version of DMET-Analyzer. coreSNP is a tool for the parallel association analysis of the SNPs variation of the patient and the clinical conditions of patients (e.g., the different response to treatments), computed by implementing the well known Fisher’s test. coreSNP to improve the statistical relevance of results, the Bonferroni and False Discovery Rate correctors are available. Finally, results are visualized as a heat map to give fast visual feedback, allowing users to interpret the results instantly.
- PARES (Parallel Association Rules Extractor from SNPs) [12] is a software tool for the parallel extraction of association rules, through a multi-thread version of an customised version of the Frequent Pattern Growth (FP-Growth) algorithm. PARES comes with a simple and intuitive graphic user interface, allowing to the user with basic or none computer science skills to mine relations between SNPs and the clinical condition of patients hidden in DMET datasets.
- Cloud4SNP [13] is a Cloud-based tool for the statistical analysis of DMET dataset. It is a Cloud-based version of DMET-Analyzer that has been realized on the Cloud by using the Data Mining Cloud Framework [14], a software environment to design and execute analysis workflows on the Cloud [15]. Cloud4SNP through the Fisher’s Test allows users to test the statistical relevance of the presence of multiple SNPs per time in a class and the clinical condition of a sample.
- ADMET Predictor is a software tool to predict the 2D structure of the molecule to create high-quality models with which to produce compounds. Molecules’ structure modeling is done through a graphical user interface. ADMET Predictor is available for Unix/Linux, Mac OS, Windows operating systems. ADMET Predictor requires to purchase a license to be used. An evaluation version of the software can be obtained by full fill a request form available on the ADMET Predictor’s website http://www.simulations-plus.com/software/admetpredictor/.
- ADMET Descriptors allows to assess compounds through queries. ADMET Descriptors is part of the BIOVIA Discovery Studio software. ADMET Descriptors provides to the user through Graphical User Interface (GUI), a set of functions to predict absorption, distribution, metabolism, excretion and toxicity (ADMET) properties starting from a list of molecules. ADMET Descriptors is available for Unix/Linux, Mac OS, and Windows operating systems. To download the trial version, users have to fill the available online form. The download of the complete version of the software requires to purchase a license, and can be done at the following web address http://accelrys.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html.
- PreADMET is a web application for predicting ADME and producing drug-like library using in-silico methods. PreADMET can be freely accessed at the following web address https://preadmet.bmdrc.kr. In the same website, the PreADMET’s purchasing editions are also available. PreADMET consists of four components Molecular-Descriptor-Calculation, Drug-likeness-Prediction, ADME-Prediction, and Toxicity-prediction, and it is developed by using C and PHP programming languages.
- ADMEWORKS ModelBuilder can be used to predict different chemical and biological properties of compounds. ADMEWORKS ModelBuilder provides functions for structural analysis, statistical methods to produce predictive models, outlier detection and analytical functions for features and sample selection. ADMEWORKS ModelBuilder requires to purchase a license to be used; trial version download is possible only filling the registration form. ADMEWORKS ModelBuilder is available at the following web address http://www.fqs.pl/en/chemistry/products/admeworks-modelbuilder.
- ADMEWORKS Predictor is a Web-based application to evaluate the ADMET properties of compounds, available at http://www.fqs.pl/en/chemistry/products/admeworks-predictor web site. ADMEWORKS Predictor provides an interactive graphical 3D structure viewer through a web browser. Also, ADMEWORKS provides the users functions for retrieving, filtering and sorting data.
3. Results
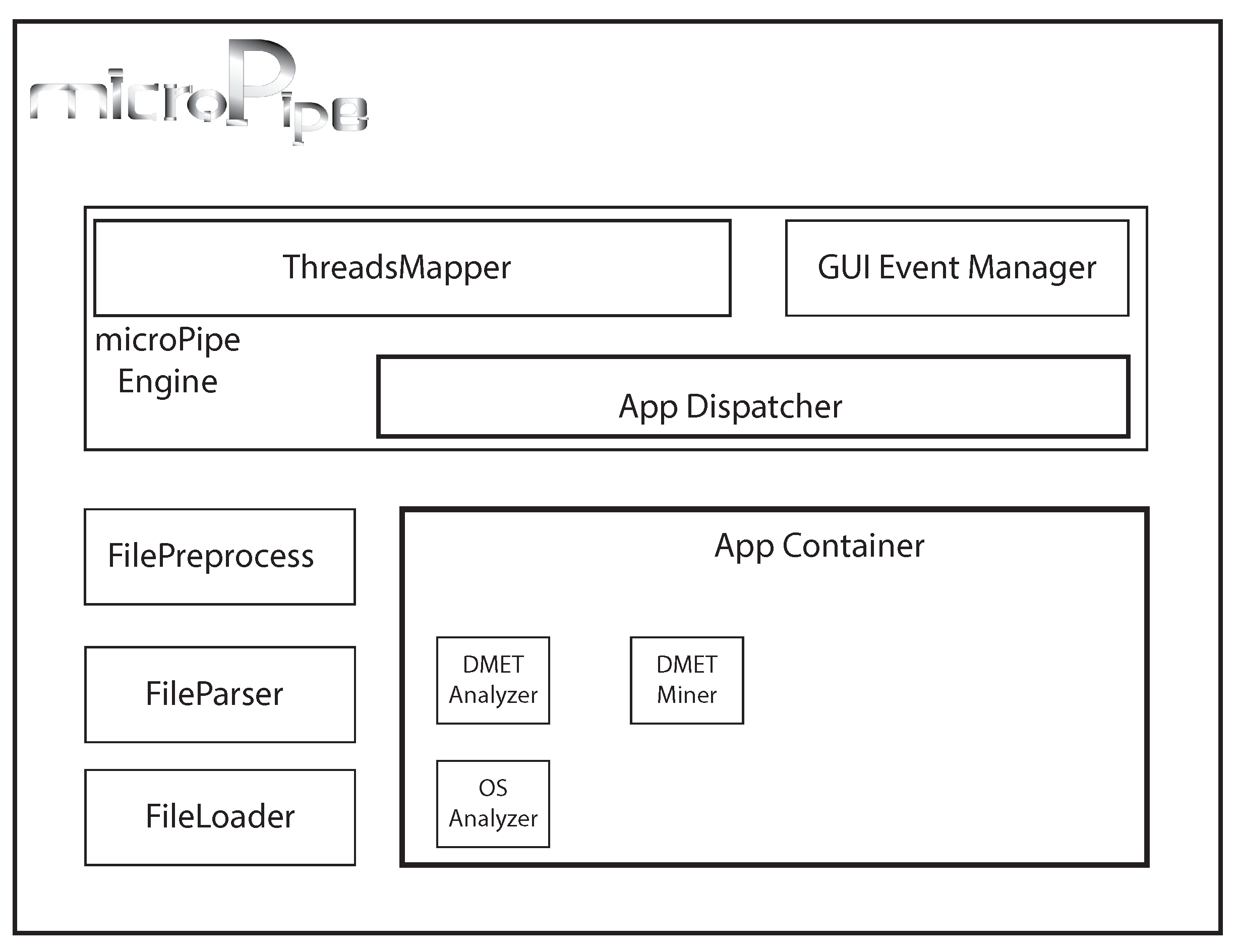
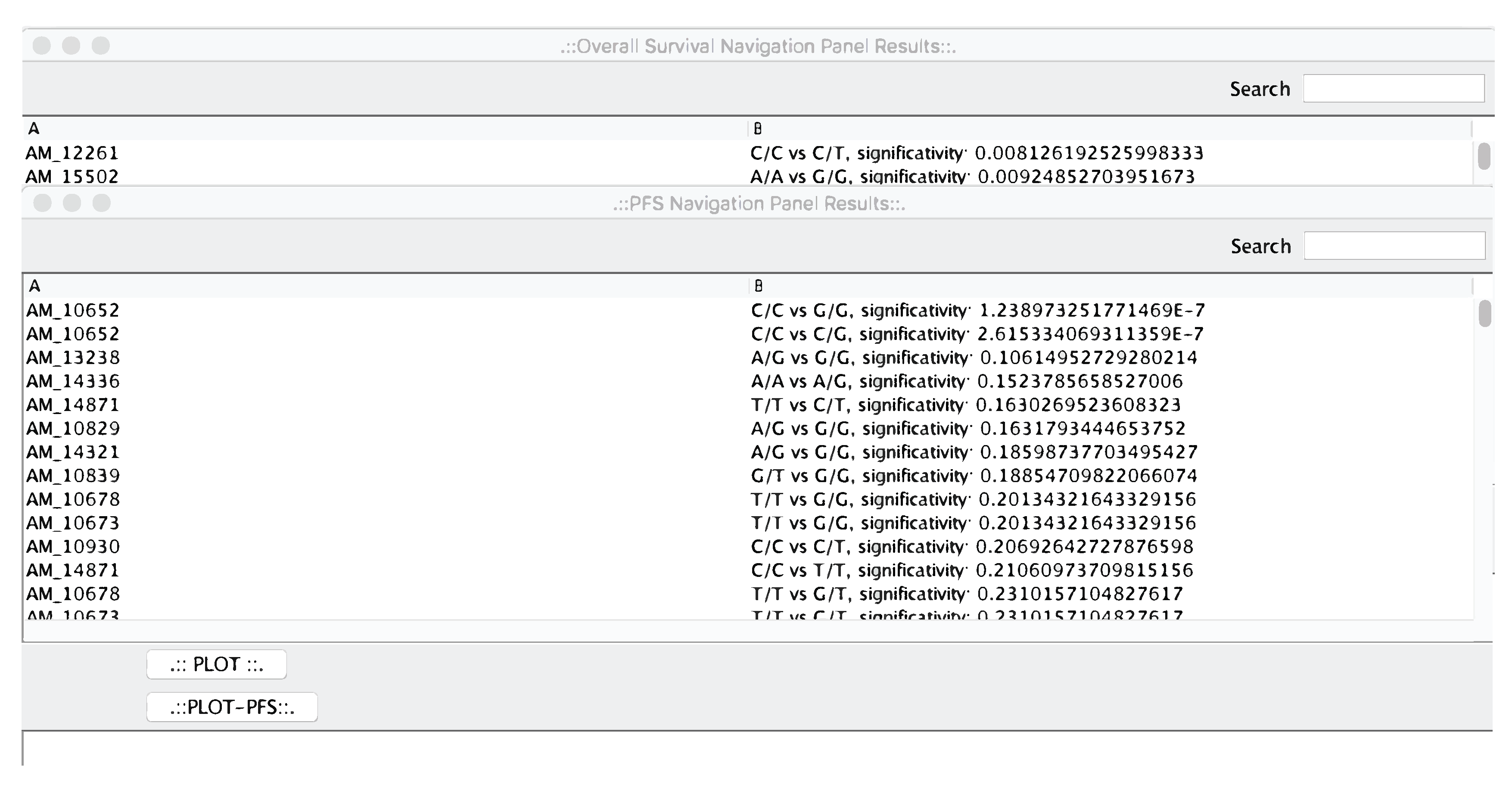
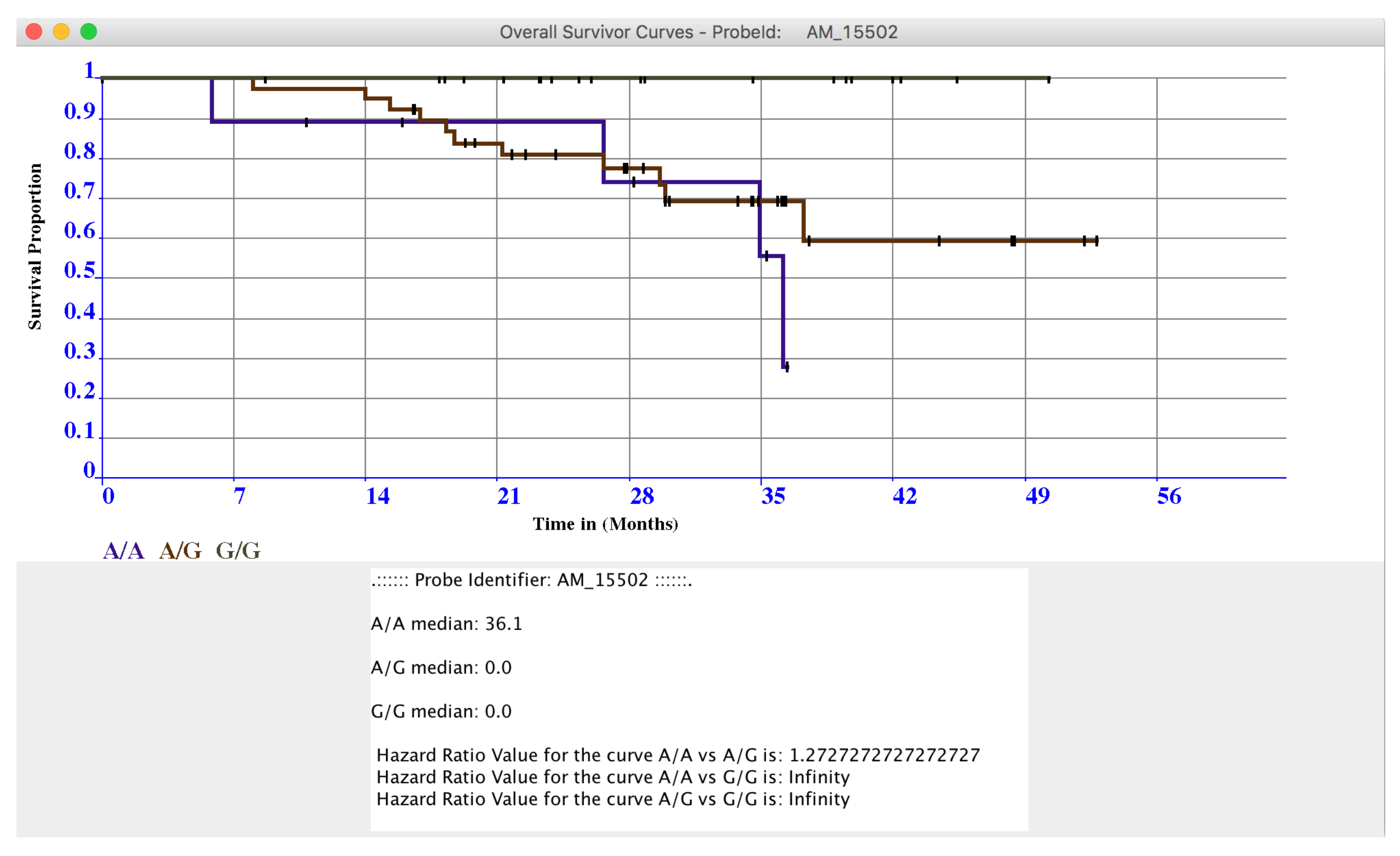
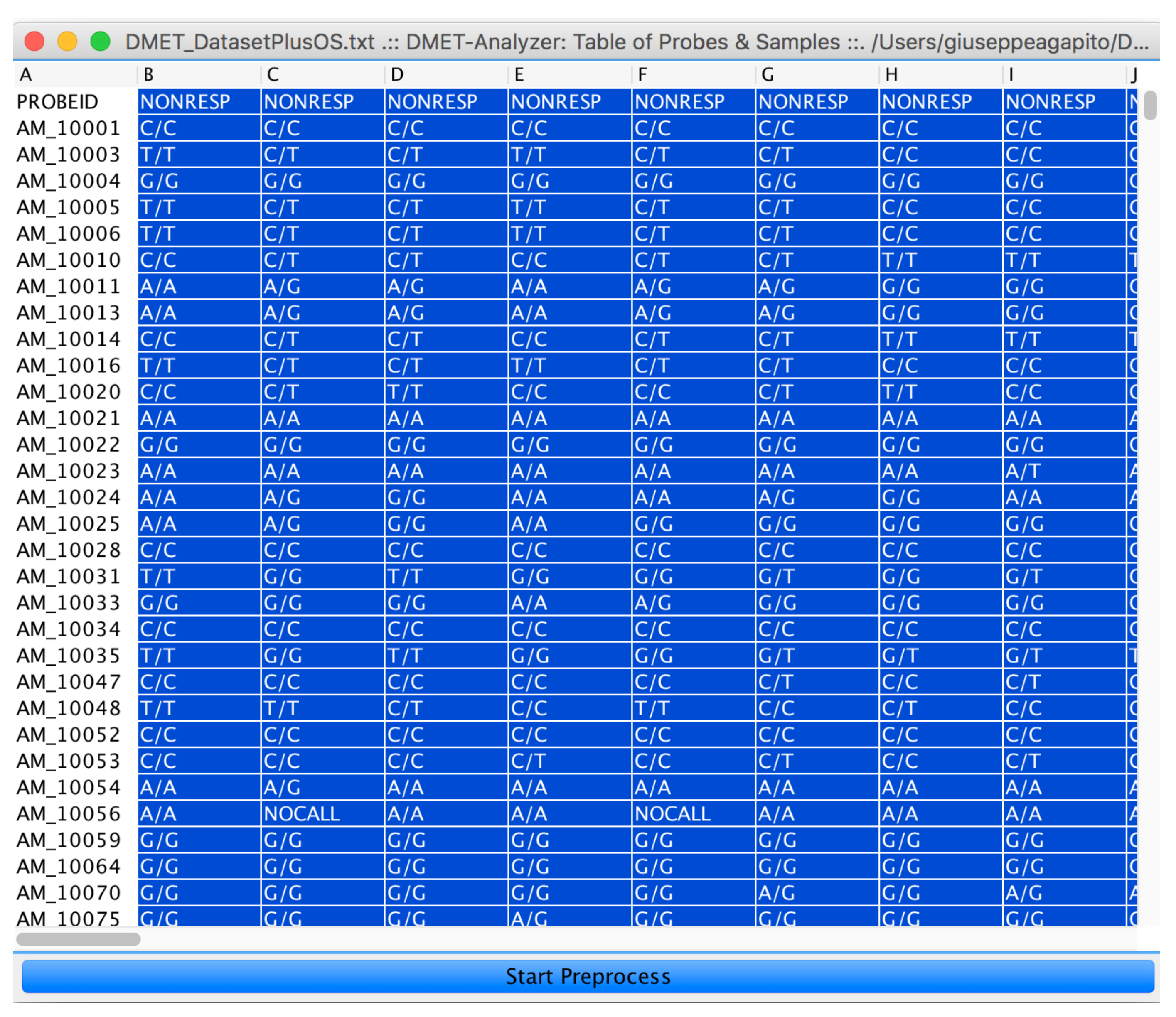
4. microPipe Usage Instruction
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| SNP | Single Nucleotide Polymorphism |
| DMET | Drug Metabolism Enzymes and Transporters |
| ADME | Adsorption, Distribution, Metabolism and Excretion |
| NGS | Next Generation Sequencing |
| GWAS | Genome-Wide Association Studies |
| MS | Mass Spectrometry |
| OS | Overall Survival |
| PFS | Progression Free Survival |
| JDK | Java Development Kit |
| jar | Java Archive |
| FDR | False Discovery Rate |
| GUI | Graphical User Interface |
| ARFF | Attribute-Relation File Format |
| CSV | Comma Separated Values |
| OSA | OS-Analyzer |
| FP-Growth | Frequent Pattern Growth |
References
- Kuipers, O.P. Genomics for food biotechnology: Prospects of the use of high-throughput technologies for the improvement of food microorganisms. Curr. Opin. Biotechnol. 1999, 10, 511–516. [Google Scholar] [CrossRef]
- Gupta, P.; Rustgi, S.; Mir, R. Array-based high-throughput DNA markers for crop improvement. Heredity 2008, 101, 5. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Tanaka, T.; Ozaki, K.; Yamada, R.; Suzuki, H.; Nakamura, Y. A high-throughput SNP typing system for genome-wide association studies. J. Hum. Genet. 2001, 46, 471. [Google Scholar] [CrossRef] [PubMed]
- Arbitrio, M.; Di Martino, M.T.; Barbieri, V.; Agapito, G.; Guzzi, P.H.; Botta, C.; Iuliano, E.; Scionti, F.; Altomare, E.; Codispoti, S.; et al. Identification of polymorphic variants associated with erlotinib-related skin toxicity in advanced non-small cell lung cancer patients by DMET microarray analysis. Cancer Chemother. Pharmacol. 2016, 77, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Arbitrio, M.; Guzzi, P.H.; Leone, E.; Baudi, F.; Piro, E.; Prantera, T.; Cucinotto, I.; Calimeri, T.; Rossi, M.; et al. A peroxisome proliferator-activated receptor gamma (PPARG) polymorphism is associated with zoledronic acid-related osteonecrosis of the jaw in multiple myeloma patients: Analysis by DMET microarray profiling. Br. J. Haematol. 2011, 154, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.T.D.; Arbitrio, M.; Leone, E.; Guzzi, P.H.; Rotundo, M.S.; Ciliberto, D.; Tomaino, V.; Fabiani, F.; Talarico, D.; Sperlongano, P.; et al. Single nucleotide polymorphisms of ABCC5 and ABCG1 transporter genes correlate to irinotecan-associated gastrointestinal toxicity in colorectal cancer patients: A DMET microarray profiling study. Cancer Biol. Ther. 2011, 12, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Arbitrio, M.; Martino, M.T.D.; Scionti, F.; Agapito, G.; Guzzi, P.H.; Cannataro, M.; Tassone, P.; Tagliaferri, P. DMET™(Drug Metabolism Enzymes and Transporters): A pharmacogenomic platform for precision medicine. Oncotarget 2016, 7, 54028–54050. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, P.H.; Agapito, G.; Di Martino, M.T.; Arbitrio, M.; Tassone, P.; Tagliaferri, P.; Cannataro, M. DMET-Analyzer: Automatic analysis of Affymetrix DMET Data. BMC Bioinform. 2012, 13, 258. [Google Scholar] [CrossRef] [PubMed]
- Agapito, G.; Guzzi, P.H.; Cannataro, M. DMET-Miner. J. Biomed. Inform. 2015, 56, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Agapito, G.; Botta, C.; Guzzi, P.H.; Arbitrio, M.; Di Martino, M.T.; Tassone, P.; Tagliaferri, P.; Cannataro, M. OSAnalyzer: A Bioinformatics Tool for the Analysis of Gene Polymorphisms Enriched with Clinical Outcomes. Microarrays 2016, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, P.H.; Agapito, G.; Cannataro, M. coreSNP: Parallel Processing of Microarray Data. IEEE Trans. Comput. 2014, 63, 2961–2974. [Google Scholar] [CrossRef]
- Agapito, G.; Guzzi, P.H.; Cannataro, M. Parallel extraction of association rules from genomics data. Appl. Math. Comput. 2017, in press. [Google Scholar] [CrossRef]
- Agapito, G.; Cannataro, M.; Guzzi, P.H.; Marozzo, F.; Talia, D.; Trunfio, P. Cloud4SNP: Distributed Analysis of SNP Microarray Data on the Cloud. In Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics, Wshington DC, USA, 22–25 September 2013; ACM: New York, NY, USA, 2013; pp. 468:468–468:475. [Google Scholar] [CrossRef]
- Marozzo, F.; Talia, D.; Trunfio, P. A Cloud Framework for Big Data Analytics Workflows on Azure; IOS Press: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Marozzo, F.; Talia, D.; Trunfio, P. Using clouds for scalable knowledge discovery applications. In Proceedings of the European Conference on Parallel Processing, Rhodes Islands, Greece, 27–31 August 2012; Springer: Berlin, Germany, 2012; pp. 220–227. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TOOL | Operating System | Statistical | Data Mining | Prediction | Modelling | Free | Pay |
|---|---|---|---|---|---|---|---|
| DMET-Analyzer | √ | × | × | × | √ | × | |
| DMET-Miner | × | √ | × | × | √ | × | |
| OS-Analyzer | √ | × | × | × | √ | × | |
| coreSNP | √ | × | × | × | √ | × | |
| PARES | × | √ | × | × | √ | × | |
| cloud4SNP | √ | × | × | × | √ | × | |
| ADMET Predictor | × | × | √ | × | × | √ | |
| ADMET Descriptors | × | × | √ | × | × | √ | |
| PreADMET | × | × | √ | × | × | √ | |
| ADMEWORKSModelB | × | × | √ | √ | × | √ | |
| ADMEWORKSPredictor | × | × | √ | × | × | √ |
| Tool | Time to Complete Analysis (s) | Used Memory (MB) |
|---|---|---|
| DMET-Analizer | 50 | 22.8 |
| DMET-Miner | 60 | 24.5 |
| OS-Analyzer | 20 | 18.7 |
| microPipe | 52 | 66.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agapito, G.; Guzzi, P.H.; Cannataro, M. A Parallel Software Pipeline for DMET Microarray Genotyping Data Analysis. High-Throughput 2018, 7, 17. https://doi.org/10.3390/ht7020017
Agapito G, Guzzi PH, Cannataro M. A Parallel Software Pipeline for DMET Microarray Genotyping Data Analysis. High-Throughput. 2018; 7(2):17. https://doi.org/10.3390/ht7020017
Chicago/Turabian StyleAgapito, Giuseppe, Pietro Hiram Guzzi, and Mario Cannataro. 2018. "A Parallel Software Pipeline for DMET Microarray Genotyping Data Analysis" High-Throughput 7, no. 2: 17. https://doi.org/10.3390/ht7020017
APA StyleAgapito, G., Guzzi, P. H., & Cannataro, M. (2018). A Parallel Software Pipeline for DMET Microarray Genotyping Data Analysis. High-Throughput, 7(2), 17. https://doi.org/10.3390/ht7020017