DMETTM Genotyping: Tools for Biomarkers Discovery in the Era of Precision Medicine

 ,
,  ,
,  ,
, 
 ,
, 
Abstract
:1. Introduction
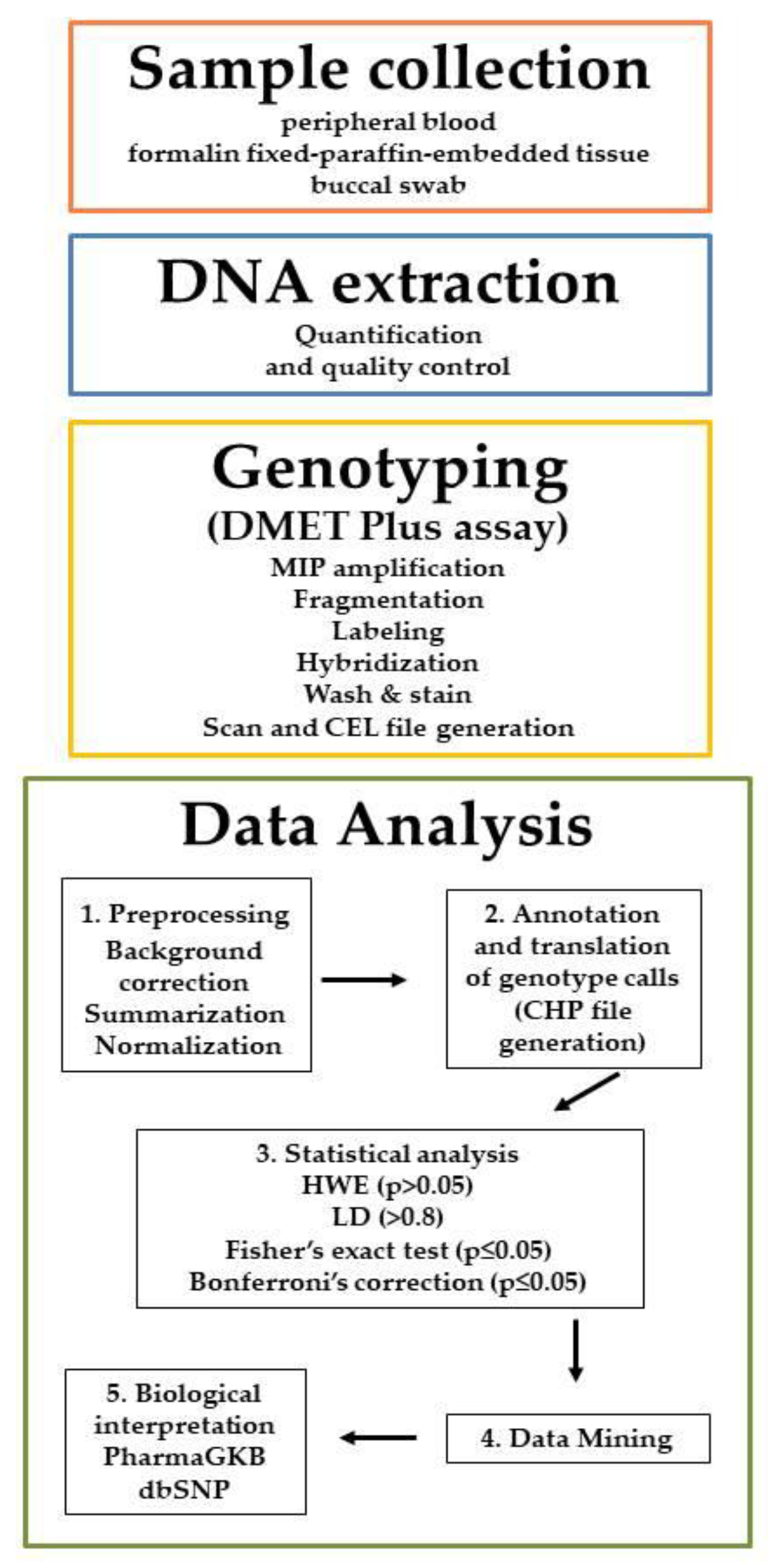
2. DMETTM Genotyping Platform
3. Software Tools to Analyze Genotyping Data
- DMET-Analyzer [22] is a software framework for the automatic association analysis between the variations present in the patients’ genome and their clinical conditions, i.e., the different response to drugs. DMET-Analyzer simplifies the identification of relevant variations into the population under investigation through the automation of the whole statistical analysis workflow. Moreover, DMET-Analyzer can annotate SNP data by automatically retrieving the information available in existing SNP databases, e.g., dbSNP, as well as to interpreter the biological process in which variants are involved through the association of SNP within a pathway, by automatically retrieving the information stored in specialized databases, e.g., PharmaGKB. DMET-Analyzer is written in Java making it compatible with Unix/Linux, MacOS and Windows operating systems, it presents a simple graphical user interface that allows non-programming users (e.g., doctors and biologists) to analyze DMET files interactively produced using the Affymetrix DMET-Console. Moreover, DMET-Analyzer implements the FDR and Bonferroni statistical correctors, the Odds-Ratio and HardyWeinberg equilibrium calculator. DMET-Analyzer can analyze only DMET files in xlsx (i.e., excels files) or plain tab delimited format (i.e., plain text files). Results can be easily saved by clicking on it and saved in textual format (txt), markup language (html) and so on. DMET-Analyzer is freely available under the GNU General Public License version 2.0 [23].
- DMET-Miner [24] is a software tool for mining association rules from DMET SNP datasets. DMET-Miner through the association rules can correlate the presence of multiple allelic variants with the clinical condition of the patients. Allowing the users to overcome the limitation of the univariate statistics implemented in DMET-Analyzer that can extract associations among a single allelic variant and the clinical conditions of samples. For example, the most frequent association among alleles responsible for the different response to a treatment. DMET-Miner enables users to automatically mine association rules from a whole DMET datasets, conversely from other available tool e.g., Weka requiring to the user to preprocess the input file in order to handle missing value, trivial data and so on. DMET-Miner is written in Java, making it available for all the operating systems compatible with Java. It presents a simple graphical user interface, allowing the users to analyze a dataset through some mouse’s clicks. DMET-Miner is distributed under Creative Commons license and is freely downloadable for academic and not-for-profit institutions [25].
- OS-Analyzer [26] is a software framework implemented in Java for the analysis of SNP microarray datasets enriched with survival events. OS-Analyzer comes with a simple, effective and intuitive graphical user interface for the automatic computation and visualization of Overall Survival (OS) and Progression Free Survival (PFS) curves of case patients, evaluating their association with ADME gene variants. Moreover, to simplify the researchers work, results according to statistical significance obtained by comparing the area under the ROC (Receiver Characteristic Curve) curves are ranked. Statistical Relevance is computed by using the log-rank test, allowing a quick and easy analysis and visualization of high-throughput data. OS-Analyzer is distributed under Creative Commons License, is freely available for academic and not-for-profit institutions [27].
- Affymetrix® DMET Console allows to preprocess the raw data file generated by the Affymetrix DMET for building a comprehensive table containing, for each probe and for each sample, the detected SNP or a No call value (i.e., ambiguous nucleotide in the SNP). DMET Console support probe-set summarization of a complete dataset of binary “.CEL” files (containing the probe-level intensities), the management of resulting preprocessed files “.CHP” (containing the gene-level information) and the building of tabular dataset containing the genotype call for all the probesets and all the samples of an experiment. Once the preprocessing phase is completed, the relationship between the detected SNPs and the response to drugs must be tested. Because of DMET Console does not allow this test, in order to discover statistically significant associations, researches have to export and manually process SNPs tables produced by the DMET Console thought the use of external tools (e.g., statistical software).
- Affymetrix Power tools suite (APT) is a set of command line programs that implement different algorithms for preprocessing Affymetrix microarray data. Two of the most popular programs in APT are: apt-dmet-genotype, for making genotype calls from Affymetrix genotyping arrays, and apt-probeset-summarize for analyzing gene expression arrays. Such as DMET Console, both programs are generally focused on .CEL file analysis.
- The GenoMetric Query Language (GMQL) [28] is a high-level query language, inspired by classic traditions of data-base management, that extends conventional algebraic operations with bioinformatics domain-specific operations specifically designed for genomics; thus, it supports knowledge discovery across thousands or even millions of samples comparing genomic regions on the basis of metric properties but also arbitrary attributes and metadata that concern regions and samples, respectively. In particular, datasets are described by the Genomic Data Model (GDM) [29], based on the notion of genomic region, which provides interoperability between several data formats. In addition, GDM combines abstractions for genomic region data with the associated experimental, biological and clinical metadata. GMQL system can be used online through a specific Web interface which provides a user-friendly intuitive environment for bioinformaticians and biologists who need to query genomic processed data (e.g., sourcing from big consortia such as ENCODE, TCGA, GENCODE or RefSeq) as well as to combine them with their private datasets (i.e., datasets created by a specific user as result of their own experiments and studies, e.g., output from Affymetrix® DMETTM Platform). Such environment provides portable and scalable genomic data management on powerful servers and clusters (based on Apache Spark).
- PLINK [30] is a free, open-source tool for GWAS and research in population genetics. PLINK works on five core functional domains: data management, summary statistics, population stratification, association analysis and identity-by-descent estimation. Association tests can be run by PLINK to evaluate case-control data to determine if an SNP has an effect on disease status. PLINK can run either as a stand-alone tool (from the command line or via shell scripting) or in conjunction with gPLINK, a Java-based graphical user interface (GUI) that offers a simple project management framework to track PLINK analyses and facilitates the integration with Haploview.
- Haploview [31] is a comprehensive suite of tools, written in Java language, for analysis and visualization of LD and haplotype maps. Haploview accepts input in a variety of formats and generates marker quality statistics, LD information, haplotype blocks, population haplotype frequencies and single marker association statistics. Haploview is fully compatible with data dumps from the HapMap project. HapMap genome browser allows researchers to explore a particular region of the genome and extract HapMap genotype data for all genotyped markers in the selected region in a format accepted by Haploview. Haploview currently supports visualization and plotting of PLINK whole genome association results [32].
- coreSNP [33] is a tool implemented in Java language, for the parallel pre-processing and statistical analysis of DMET SNP datasets. The scalable implementation is obtained exploiting the multi-threading capabilities of modern CPUs, allowing core SNP to manage huge amount of DMET data. The automatic association analysis between possible genome variations of the patient and the clinical conditions through the well-known Fisher’s Test is obtained. Moreover, multiple-statistical correctors i.e., Bonferroni and False Discovery Rate, with which to improve the statistical significance of results, are available. The visualization of the detected SNPs as heatmap plot provides a visual feedback that simplifies the interpretation of the results.
- Parallel Association Rules Extractor from SNPs (PARES) [34] is a multi-thread software tool developed in Java for the parallel extraction of association rules by which to correlate the presence of a multiple allelic variants with the patients’ clinical condition, i.e., the most likely set of alleles responsible for the onset of adverse drug reactions. PARES is a multi-thread version based on the optimized version of the Frequent Pattern Growth (FP-Growth) algorithm. PARES encompasses a customized SNP dataset preprocessing approach based on a Fisher’s Test Filter to prune trivial transactions, allowing to shrink the search space as well as to reduce the FP-Tree size enabling a better management of the main memory. PARES comes with a simple and intuitive graphic user interface, where specific skills are not necessary to extract multiple relations between genomic factors buried into the datasets. PARES is distributed by BioinfoLabUnicz, under Creative Commons license, is freely downloadable for academic and not-for-profit institutions [35].
- GenotypeAnalytics [36] is a representational state transfer (RESTFul) service by which to mine association rules from SNP-dataset through the use of a common web-browser. GenotypeAnalytics can speed up and simplify the analysis of massive amount of SNPs data, highlighting in a remarkable way only the SNP involved in the development of the disease or in adverse drug reaction. GenotypeAnalytics can support researcher and medical doctors to discern new molecular markers that can be (after further opportune validations) used into the clinical contexts. Possible new molecular markers can be used to perform pathway enrichment to understand which SNPs are responsible for the pathway’s anomalies or the SNPs that influence drug responses in subjects with the same pathology following the same therapy.
- Cloud4SNP [37] is a Cloud software tool for the parallel pre-processing and statistical analysis of DMET SNPs data. Cloud4SNP is the Cloud version of DMET-Analyzer [22] that has been implemented on the Cloud using the Data Mining Cloud Framework, a software environment for the design and the execution of knowledge discovery workflows on the Cloud. Cloud4SNP is developed in Java and presents a simple graphical user interface that can be accessed by means of a common web-browser. Providing the analysis workflow as a service, it allows the users to upload and analyze the data without to buy expensive hardware or to setup the analysis environment. Cloud4SNP allows one to identify the relevance of the presence of SNPs in one of the two classes of samples using the well-known Fisher test, along with the use of multiple-statistical correctors such as Bonferroni and FDR.
4. A New Methodology to Integrate Genomic Public Data with Genotyping Private Data
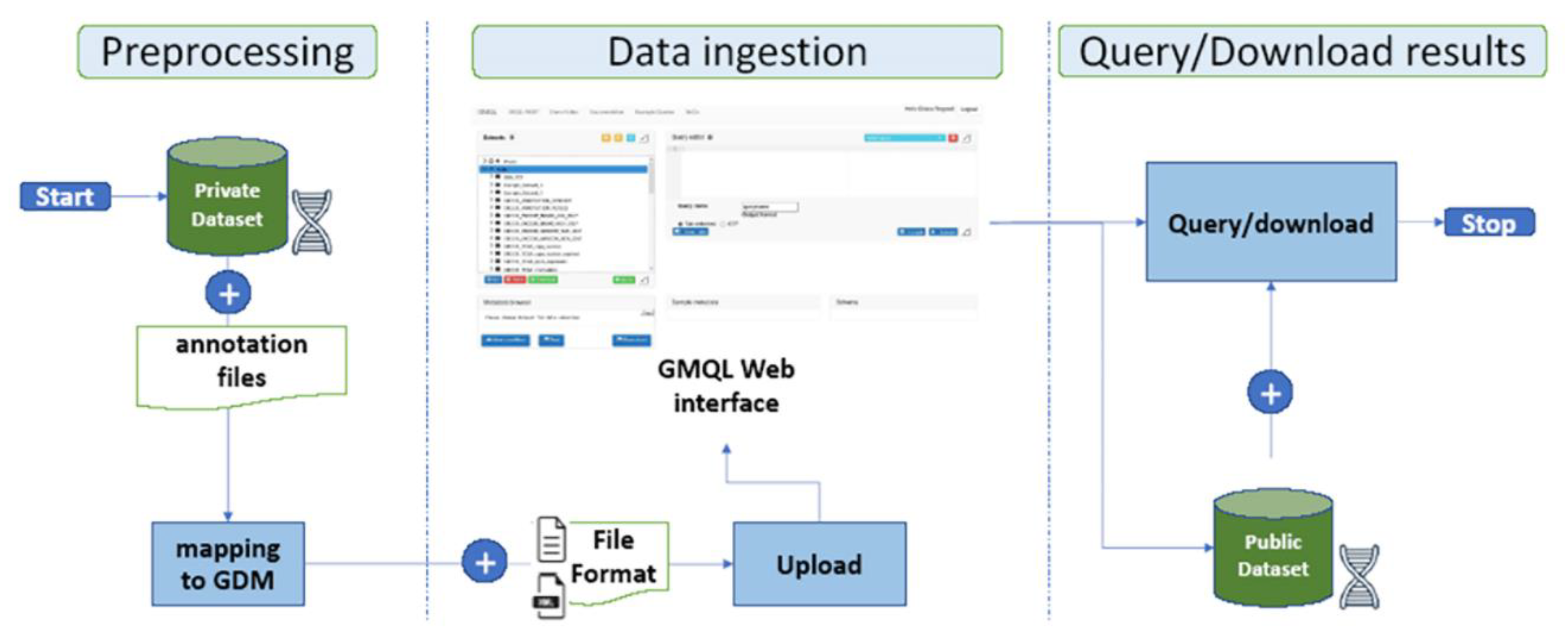
4.1. Data Preprocessing Stage
4.2. Data Ingestion Stage
- the standard file-format mode allows one to use a number of file standard formats directly supported by the system [39] (e.g., BED, NarrowPeak, BroadPeak, VCF, etc);
- the custom file-format mode can be chosen by selecting the “Custom (GTF or tab/delimited)” option and it allows one to use a user-defined format following the guidelines of Gene Transfer Format (GTF) [40] or TAB-delimited formats. In this case it is required the definition of an additional Extended Markup Language (XML) format Schema file describing the structure of the dataset to upload. Once uploaded, the private datasets are shown in the “interface datasets viewer” under the “Private” folder and it can be managed independently or in combination with other public datasets.
4.3. Query/Download Results Stage
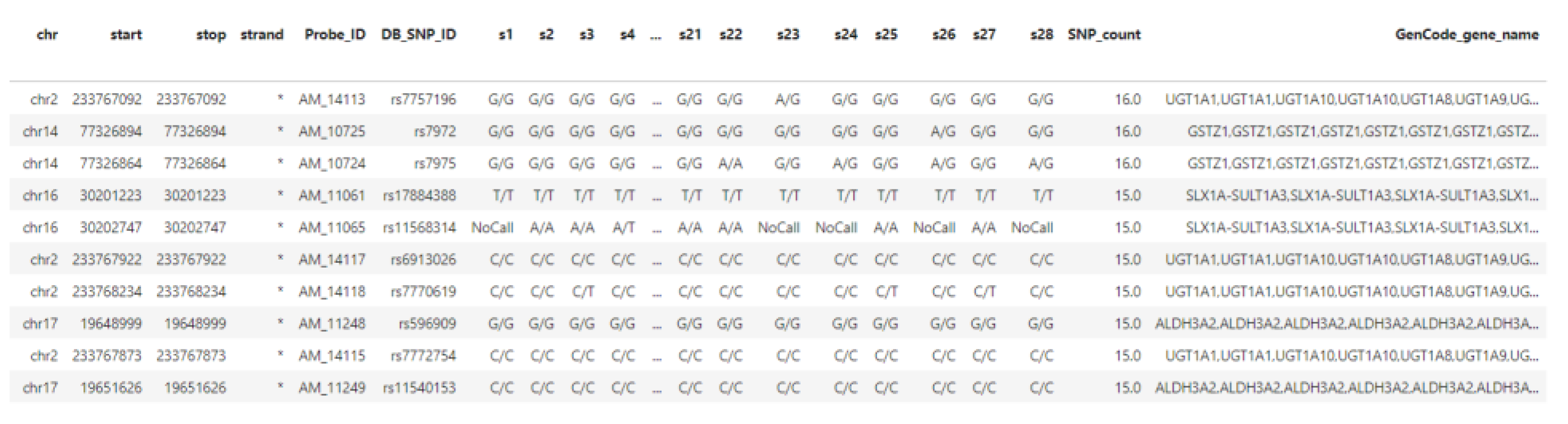
5. Case Study
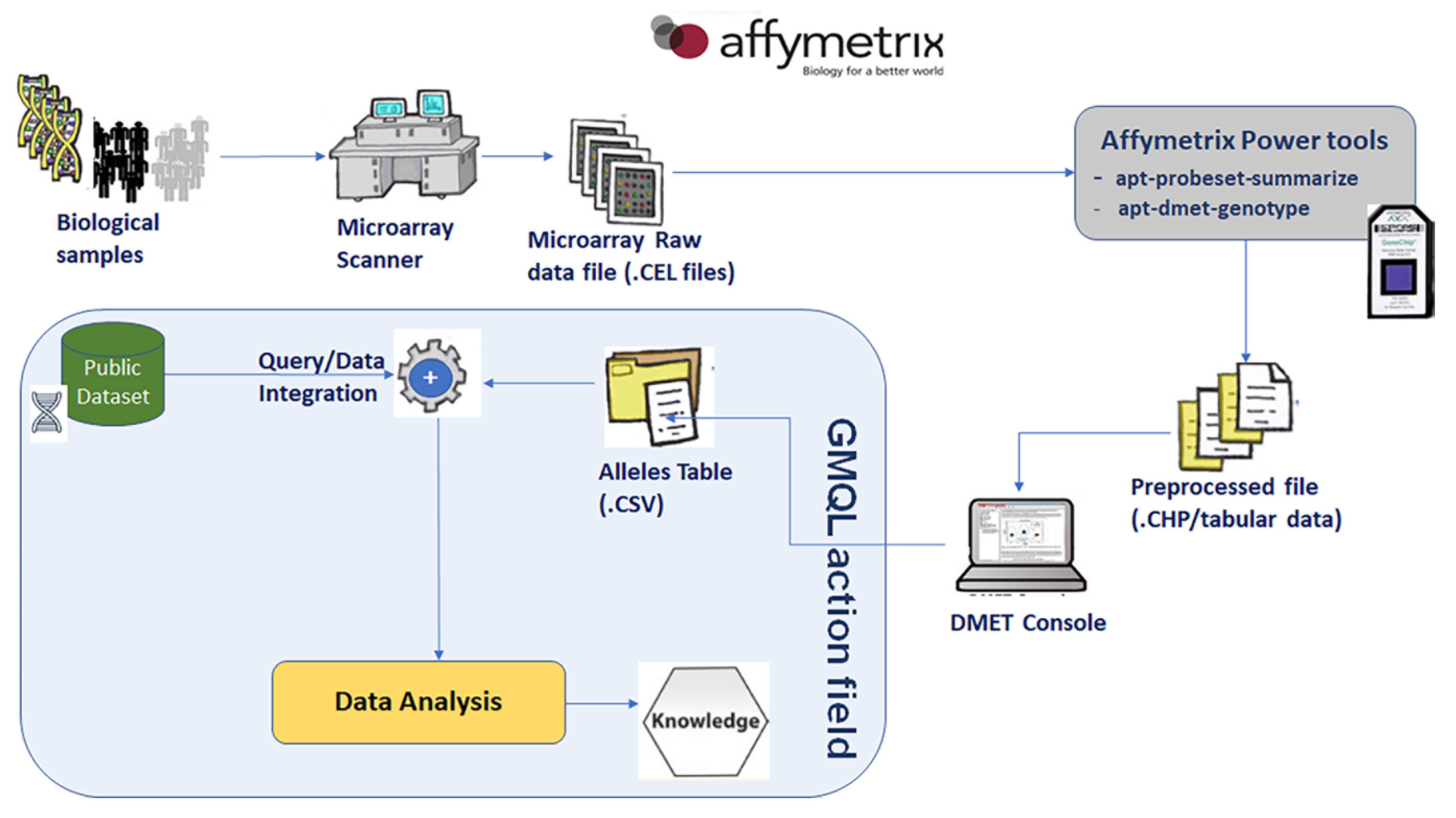
5.1. Integration of Private DMET Platform Data with GENCODE Annotation Database
5.2. Data Preprocessing Stage
5.3. Data Ingestion
5.4. Query/Download Results
- select SNPs from DMET Dataset;
- select exon regions from GENCODE annotation dataset;
- map exon regions on SNPs regions;
- select only SNPs regions that overlap at least one exon region.
6. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Southey, M.C.; Goldgar, D.E.; Winqvist, R.; Pylkas, K.; Couch, F.; Tischkowitz, M.; Foulkes, W.D.; Dennis, J.; Michailidou, K.; van Rensburg, E.J.; et al. Palb2, chek2 and atm rare variants and cancer risk: Data from cogs. J. Med. Genet. 2016, 53, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Crawford, B.; Adams, S.B.; Sittler, T.; van den Akker, J.; Chan, S.; Leitner, O.; Ryan, L.; Gil, E.; van ’t Veer, L. Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast and ovarian cancer patients. Breast Cancer Res. Treat. 2017, 163, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Tagliaferri, P.; Ventura, M.; Baudi, F.; Cucinotto, I.; Arbitrio, M.; Di Martino, M.T.; Tassone, P. Brca1/2 genetic background-based therapeutic tailoring of human ovarian cancer: Hope or reality? J. Ovarian Res. 2009, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Zhou, Z.; Zhou, J.; Chen, S.Q. Pharmacogenomics of drug metabolizing enzymes and transporters: Relevance to precision medicine. Genom. Proteom. Bioinform. 2016, 14, 298–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.S.; Ratain, M.J. Pharmacogenetics and pharmacogenomics of anticancer agents. CA Cancer J. Clin. 2009, 59, 42–55. [Google Scholar] [CrossRef]
- Judson, R.; Stephens, J.C.; Windemuth, A. The predictive power of haplotypes in clinical response. Pharmacogenomics 2000, 1, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Motsinger-Reif, A.A.; Jorgenson, E.; Relling, M.V.; Kroetz, D.L.; Weinshilboum, R.; Cox, N.J.; Roden, D.M. Genome-wide association studies in pharmacogenomics: Successes and lessons. Pharm. Genom. 2013, 23, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, U.I.; Gulilat, M.; Kim, R.B. The role of next-generation sequencing in pharmacogenetics and pharmacogenomics. Cold Spring Harb. Perspect. Med. 2019, 9, a033027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbitrio, M.; Di Martino, M.T.; Scionti, F.; Barbieri, V.; Pensabene, L.; Tagliaferri, P. Pharmacogenomic profiling of adme gene variants: Current challenges and validation perspectives. High-throughput 2018, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Di Martino, M.T.; Arbitrio, M.; Leone, E.; Guzzi, P.H.; Rotundo, M.S.; Ciliberto, D.; Tomaino, V.; Fabiani, F.; Talarico, D.; Sperlongano, P.; et al. Single nucleotide polymorphisms of abcc5 and abcg1 transporter genes correlate to irinotecan-associated gastrointestinal toxicity in colorectal cancer patients: A dmet microarray profiling study. Cancer Biol. Ther. 2011, 12, 780–787. [Google Scholar] [CrossRef]
- Di Martino, M.T.; Arbitrio, M.; Guzzi, P.H.; Leone, E.; Baudi, F.; Piro, E.; Prantera, T.; Cucinotto, I.; Calimeri, T.; Rossi, M.; et al. A peroxisome proliferator-activated receptor gamma (pparg) polymorphism is associated with zoledronic acid-related osteonecrosis of the jaw in multiple myeloma patients: Analysis by dmet microarray profiling. Br. J. Haematol. 2011, 154, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scionti, F.; Di Martino, M.T.; Sestito, S.; Nicoletti, A.; Falvo, F.; Roppa, K.; Arbitrio, M.; Guzzi, P.H.; Agapito, G.; Pisani, A.; et al. Genetic variants associated with fabry disease progression despite enzyme replacement therapy. Oncotarget 2017, 8, 107558–107564. [Google Scholar] [CrossRef] [Green Version]
- Di Martino, M.T.; Scionti, F.; Sestito, S.; Nicoletti, A.; Arbitrio, M.; Hiram Guzzi, P.; Talarico, V.; Altomare, F.; Sanseviero, M.T.; Agapito, G.; et al. Genetic variants associated with gastrointestinal symptoms in fabry disease. Oncotarget 2016, 7, 85895–85904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbitrio, M.; Di Martino, M.T.; Barbieri, V.; Agapito, G.; Guzzi, P.H.; Botta, C.; Iuliano, E.; Scionti, F.; Altomare, E.; Codispoti, S.; et al. Identification of polymorphic variants associated with erlotinib-related skin toxicity in advanced non-small cell lung cancer patients by dmet microarray analysis. Cancer Chemother. Pharmacol. 2016, 77, 205–209. [Google Scholar] [CrossRef]
- Arbitrio, M.; Scionti, F.; Altomare, E.; Di Martino, M.T.; Agapito, G.; Galeano, T.; Staropoli, N.; Iuliano, E.; Grillone, F.; Fabiani, F.; et al. Polymorphic variants in nr1i3 and ugt2b7 predict taxane neurotoxicity and have prognostic relevance in patients with breast cancer: A case-control study. Clin. Pharmacol. Ther. 2019, 106, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Deeken, J. The affymetrix dmet platform and pharmacogenetics in drug development. Curr. Opin. Mol. Ther. 2009, 11, 260–268. [Google Scholar] [PubMed]
- Hardenbol, P.; Yu, F.L.; Belmont, J.; MacKenzie, J.; Bruckner, C.; Brundage, T.; Boudreau, A.; Chow, S.; Eberle, J.; Erbilgin, A.; et al. Highly multiplexed molecular inversion probe genotyping: Over 10,000 targeted snps genotyped in a single tube assay. Genome. Res. 2005, 15, 269–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, C.A.; Smith, C.; Yang, W.; Lorier, R.; Crews, K.R.; Kornegay, N.; Hicks, J.K.; Stewart, C.F.; Kawedia, J.D.; Ramsey, L.B.; et al. Concordance of dmet plus genotyping results with those of orthogonal genotyping methods. Clin. Pharmacol. Ther. 2012, 92, 360–365. [Google Scholar] [CrossRef]
- Arbitrio, M.; Di Martino, M.T.; Scionti, F.; Agapito, G.; Guzzi, P.H.; Cannataro, M.; Tassone, P.; Tagliaferri, P. Dmet (drug metabolism enzymes and transporters): A pharmacogenomic platform for precision medicine. Oncotarget 2016, 7, 54028–54050. [Google Scholar] [CrossRef]
- Guzzi, P.H.; Agapito, G.; Milano, M.; Cannataro, M. Methodologies and experimental platforms for generating and analysing microarray and mass spectrometry-based omics data to support p4 medicine. Brief. Bioinform. 2016, 17, 553–561. [Google Scholar] [CrossRef]
- Settino, M.; Cannataro, M. Survey of main tools for querying and analyzing tcga data. In Proceedings of the 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Madrid, Spain, 3–6 December 2018; pp. 1711–1718. [Google Scholar]
- Guzzi, P.H.; Agapito, G.; Di Martino, M.T.; Arbitrio, M.; Tassone, P.; Tagliaferri, P.; Cannataro, M. Dmet-analyzer: Automatic analysis of affymetrix dmet data. BMC Bioinform. 2012, 13, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DMET-Analyzer. Available online: https://sourceforge.net/projects/dmetanalyzer/ (accessed on 26 March 2020).
- Agapito, G.; Guzzi, P.H.; Cannataro, M. Dmet-miner: Efficient discovery of association rules from pharmacogenomic data. J. Biomed. Inform. 2015, 56, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DMET-Miner. Available online: https://sourceforge.net/projects/dmetanalyzer/ (accessed on 26 March 2020).
- Agapito, G.; Botta, C.; Guzzi, P.H.; Arbitrio, M.; Di Martino, M.T.; Tassone, P.; Tagliaferri, P.; Cannataro, M. Osanalyzer: A bioinformatics tool for the analysis of gene polymorphisms enriched with clinical outcomes. Microarrays 2016, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OS-Analyzer. Available online: https://sites.google.com/site/overallsurvivalanalyzer/ (accessed on 26 March 2020).
- Masseroli, M.; Pinoli, P.; Venco, F.; Kaitoua, A.; Jalili, V.; Palluzzi, F.; Muller, H.; Ceri, S. Genometric query language: A novel approach to large-scale genomic data management. Bioinformatics 2015, 31, 1881–1888. [Google Scholar] [CrossRef]
- Masseroli, M.; Kaitoua, A.; Pinoli, P.; Ceri, S. Modeling and interoperability of heterogeneous genomic big data for integrative processing and querying. Methods 2016, 111, 3–11. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- gPLINK. Available online: http://zzz.bwh.harvard.edu/plink/gplink.shtml (accessed on 26 March 2020).
- Guzzi, P.H.; Agapito, G.; Cannataro, M. Coresnp: Parallel processing of microarray data. IEEE Trans. Comput. 2014, 63, 2961–2974. [Google Scholar] [CrossRef]
- Agapito, G.; Guzzi, P.H.; Cannataro, M. Parallel extraction of association rules from genomics data. Appl. Math. Comput. 2019, 350, 434–446. [Google Scholar] [CrossRef]
- PARES. Available online: https://sites.google.com/site/pareswebsite/. (accessed on 26 March 2020).
- Agapito, G.; Guzzi, P.H.; Cannataro, M. Using genotypeanalytics to analyze pharmacogenomic datasets. Acm-Bcb’ 2017. In Proceedings of the 8th Acm International Conference on Bioinformatics, Computational Biology, and Health Informatics. Association for Computing Machinery, New York NY, USA, 20–23 August 2017; pp. 740–743. [Google Scholar]
- Agapito, G.; Cannataro, M.; Guzzi, P.H.; Marozzo, F.; Talia, D.; Trunfio, P. Cloud4snp: Distributed analysis of snp microarray data on the cloud. In ACM Conference on Bioinformatics, Computational Biology and Biomedical Informatics, ACM-BCB 2013; Association for Computing Machinery: New York NY, USA, September 2013; pp. 468–475. [Google Scholar]
- Marozzo, F.; Talia, D.; Trunfio, P. A cloud framework for big data analytics workflows on azure. Adv. Parallel Comput. 2013, 23, 182–191. [Google Scholar]
- Goossens, N.; Nakagawa, S.; Sun, X.C.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [PubMed]
- Standard File Format. Available online: https://github.com/DEIB-GECO/GMQL-WEB/wiki/file_formats (accessed on 26 March 2020).
- GTF. Available online: http://mblab.wustl.edu/GTF22.html (accessed on 26 March 2020).
- DDGC. Available online: http://www.bioinformatics.deib.polimi.it/geco/?try (accessed on 26 March 2020).
- GENCODE database. Available online: https://www.gencodegenes.org/ (accessed on 26 March 2020).
- DMET Annotations. Available online: https://www.affymetrix.com/support/developer/powertools/changelog/VIGNETTE-DMET-genotyping.html (accessed on 26 March 2020).
- UCSC Lift Genome Annotations Tool. Available online: https://genome.ucsc.edu/cgi-bin/hgLiftOver (accessed on 26 March 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | Availability | Main Features | IA | Interface | OS | DM | Statistical | Prep | CS | Weaknesses | Strengths |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DMET-Analyzer | Free | Analysis of Variants based on Statistical test | Annotations and direct link to external databases | Command line, GUI | Platform-independent | NO | YES | YES | L | Single allelic variants discovery; Unable to dig with Affymetrix raw files; | It automatically analyses data in case-control association studies; |
| DMET-Miner | Free | Analysis of Variants using Association Rules mining | Annotations and direct link to external databases | GUI | Platform-independent | YES | NO | YES | L | Manage small Dataset | Multiple allelic variants discovery |
| OS-Analyzer | Free | computation and visualization of OS and PFS curves | Integration of genomic and clinical data | GUI | Platform-independent | NO | YES | YES | L | It cannot analyse gene expression data in order to plot OS curves (3) | High performance with respect to other statistical tools; Automatic analysis of whole DMET SNPs Dataset |
| Affymetrix DMET-Console | Free* | Preprocessing of raw data generated by DMET | NO | GUI | Windows | NO | NO | YES | L | Lacks in the possibility of doing statistical and data mining analysis; | It allows only the preprocessing of binary data |
| Affymetrix Power tools suite (APT) | Free* | They implement algorithms for analyzing and working with Affymetrix Microarrays | YES | command line | Platform-independent | NO | NO | YES | M | Lacks in the possibility of doing statistical and data mining analysis | Analysis of intensity microarray data to produce final tabular dataset |
| The GenoMetric Query Language (GMQL) | Free | Query, Download and Integrate Public with Private Genomic Datasets | Integrate Public with Private Genomic Datasets | Web interface | Platform-independent | NO | NO | NO | M | Allows to perform only genometric queries | It combines private dataset with publicly available datasets |
| PLINK | Free | Analysis of genotype/phenotype data | YES | command line, GUI (5) | Platform-independent | NO | YES | YES | H | Allows to perform only statistical analysis | Data management, statistical analysis, association analysis of whole-genome studies |
| Haploview | Free | Haplotype analysis | YES | GUI | Platform-independent | NO | YES | NO | M | Allows to perform only statistical analysis | Graphical computation of LD statistics and population haplotype patterns |
| coreSNP | Free | Parallel analysis of Variants based on Statistical test | NO | GUI | Platform-independent | NO | YES | YES | L | single allelic variants discovery; Unable to dig with Affymetrix raw files | Massive parallel analysis of SNPs dataset |
| PARES | Free | Parallel association rules extraction from SNP Datasets | NO | GUI | Platform-independent | YES | NO | YES | L | Manage small Dataset | Multiple allelic variants discovery |
| Genotype Analytics | Free | Web Services for bioinformatics | YES | Web interface | Platform-independent | YES | YES | YES | L | Data mining and statistical Web Services to analyse SNP Datasets | |
| Cloud4SNP | Free | Cloud Serices to analyse SNP Datasets | YES | Web interface | Platform-independent | NO | YES | YES | L | Allows to perform only statistical analysis | Easy to use through Web Browser |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agapito, G.; Settino, M.; Scionti, F.; Altomare, E.; Guzzi, P.H.; Tassone, P.; Tagliaferri, P.; Cannataro, M.; Arbitrio, M.; Di Martino, M.T. DMETTM Genotyping: Tools for Biomarkers Discovery in the Era of Precision Medicine. High-Throughput 2020, 9, 8. https://doi.org/10.3390/ht9020008
Agapito G, Settino M, Scionti F, Altomare E, Guzzi PH, Tassone P, Tagliaferri P, Cannataro M, Arbitrio M, Di Martino MT. DMETTM Genotyping: Tools for Biomarkers Discovery in the Era of Precision Medicine. High-Throughput. 2020; 9(2):8. https://doi.org/10.3390/ht9020008
Chicago/Turabian StyleAgapito, Giuseppe, Marzia Settino, Francesca Scionti, Emanuela Altomare, Pietro Hiram Guzzi, Pierfrancesco Tassone, Pierosandro Tagliaferri, Mario Cannataro, Mariamena Arbitrio, and Maria Teresa Di Martino. 2020. "DMETTM Genotyping: Tools for Biomarkers Discovery in the Era of Precision Medicine" High-Throughput 9, no. 2: 8. https://doi.org/10.3390/ht9020008
APA StyleAgapito, G., Settino, M., Scionti, F., Altomare, E., Guzzi, P. H., Tassone, P., Tagliaferri, P., Cannataro, M., Arbitrio, M., & Di Martino, M. T. (2020). DMETTM Genotyping: Tools for Biomarkers Discovery in the Era of Precision Medicine. High-Throughput, 9(2), 8. https://doi.org/10.3390/ht9020008