Composition-Dependent Sorptive Fractionation of Anthropogenic Dissolved Organic Matter by Fe(III)-Montmorillonite

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sorbent Preparation
2.2. DOM Extraction and Fractionation
2.3. Sorption Experiments
2.4. FT-ICR MS Analysis
2.4.1. Sample Preparation
2.4.2. Instrumental Analysis
2.4.3. Data Analysis
3. Results
3.1. Sorption by Mass
3.2. Presence of “Surfactant-Like” Peaks
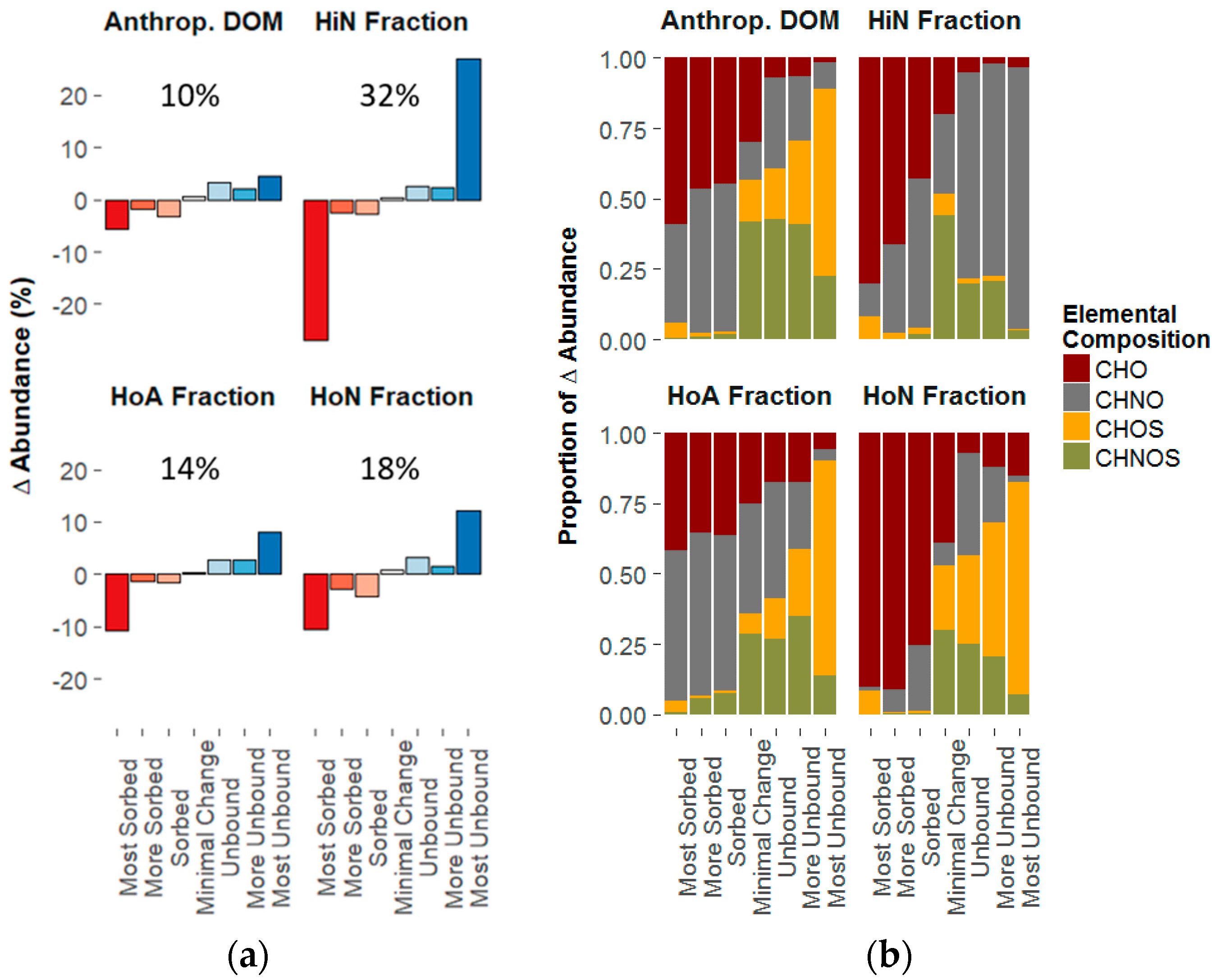
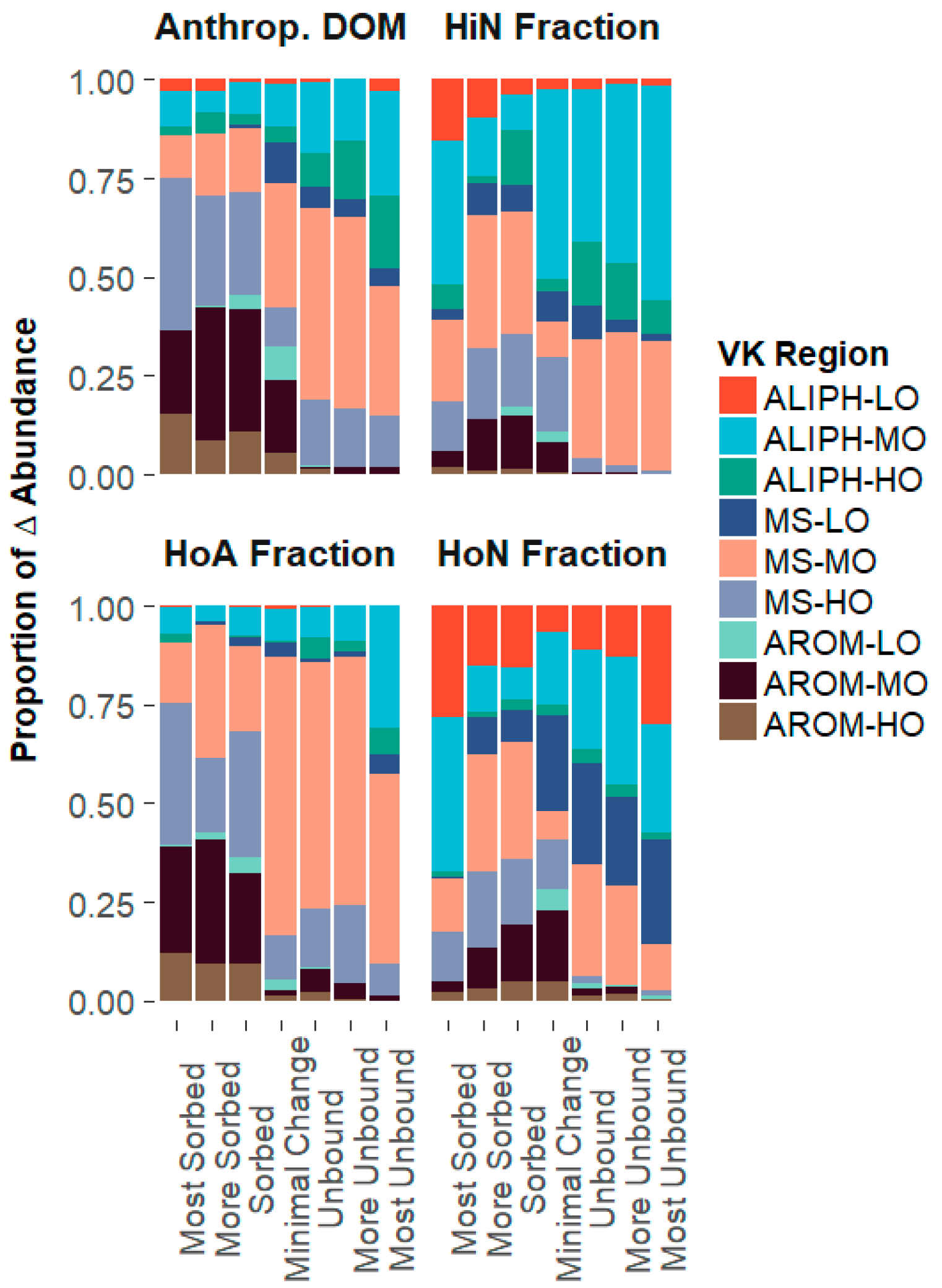
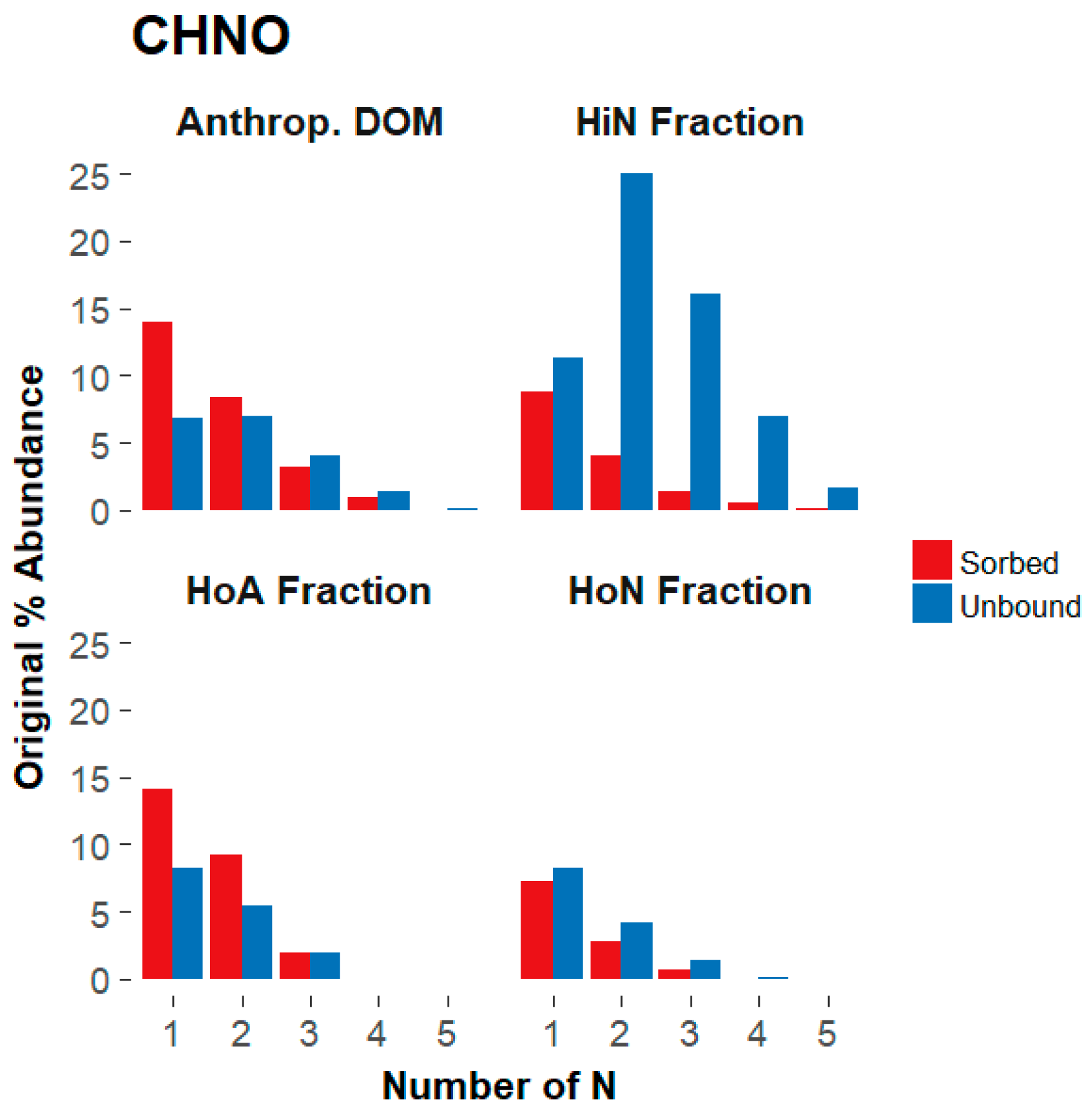
3.3. Composition-Dependent Sorptive Fractionation
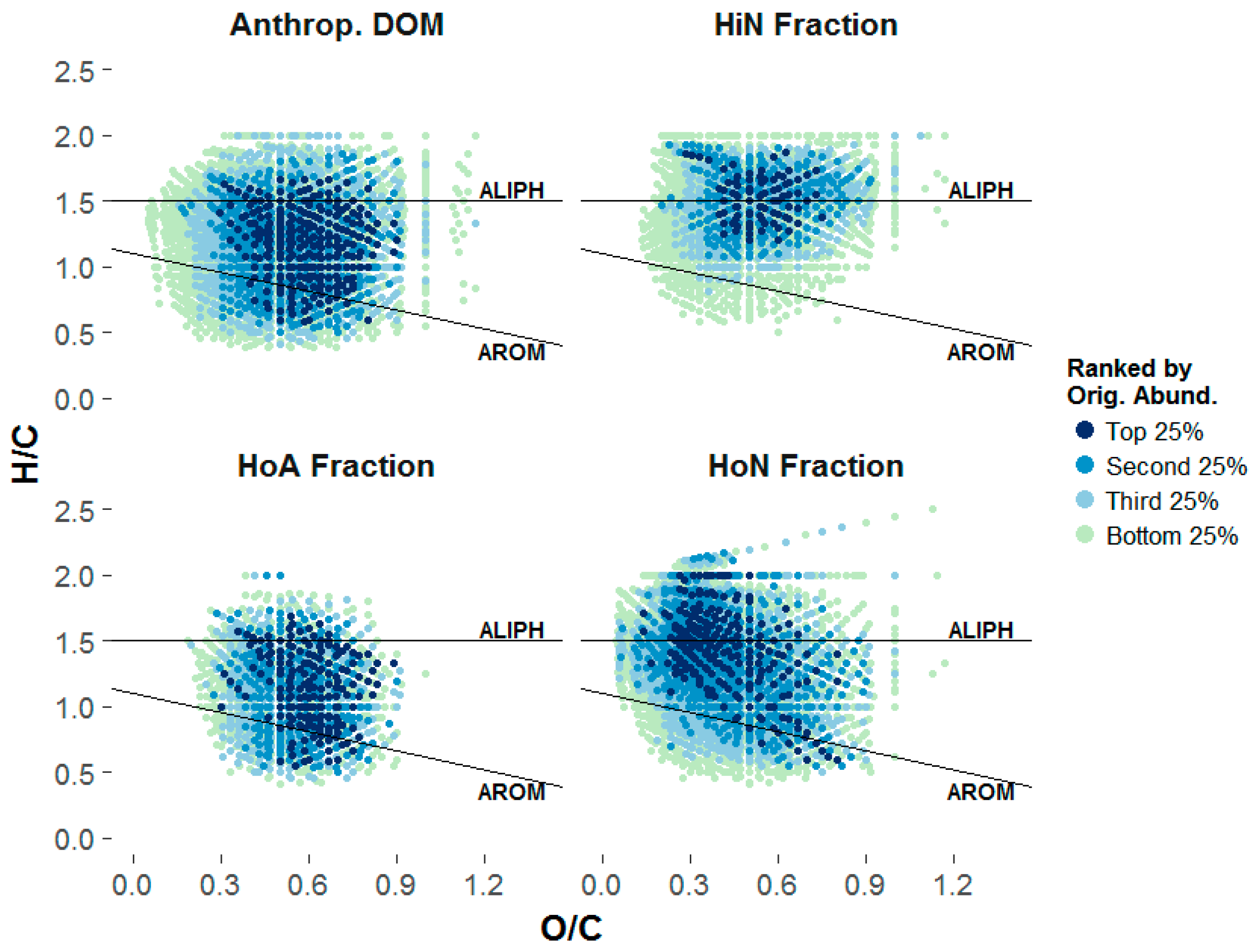
3.3.1. Composition of the Structural Fractions
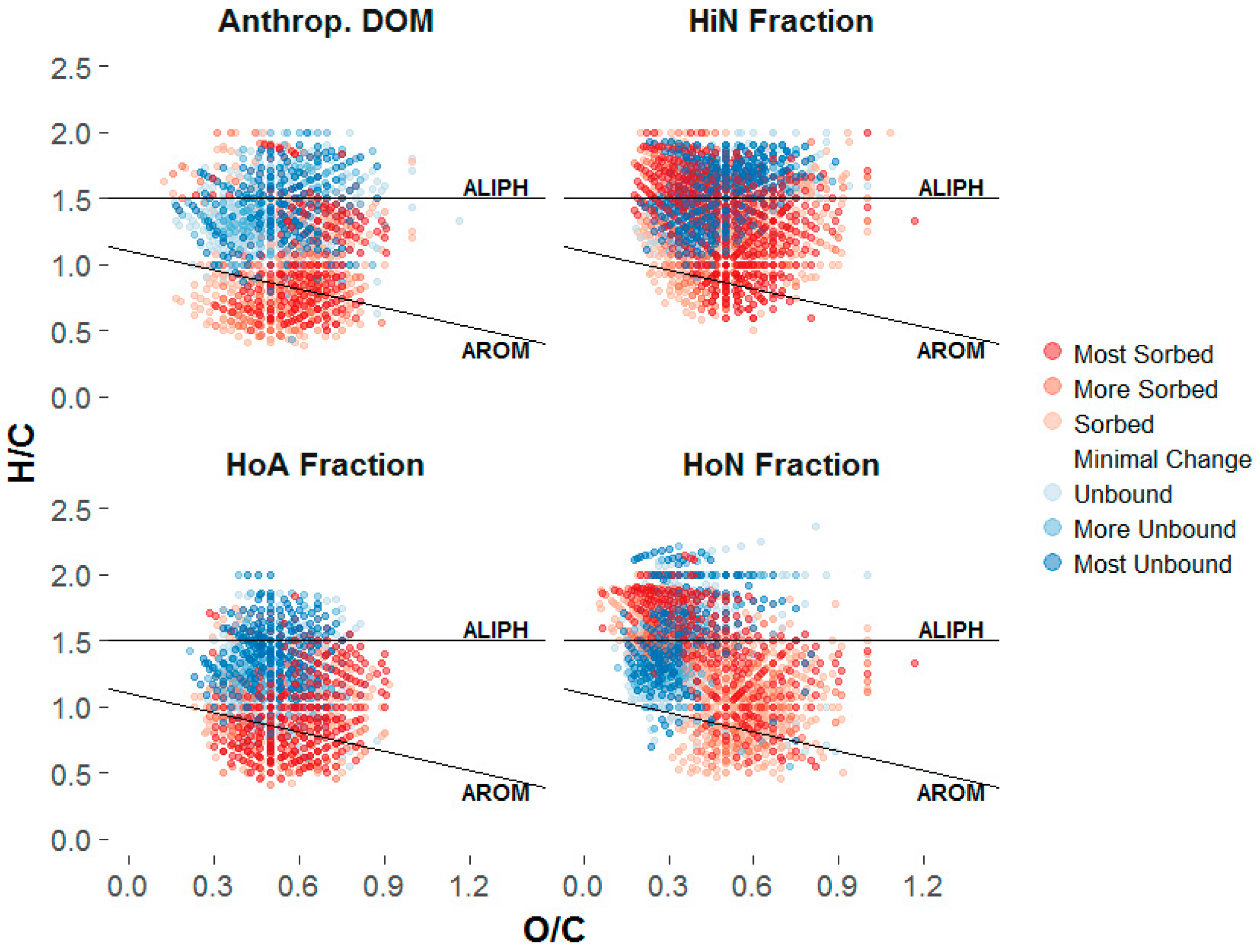
3.3.2. Sorptive Fractionation
4. Discussion
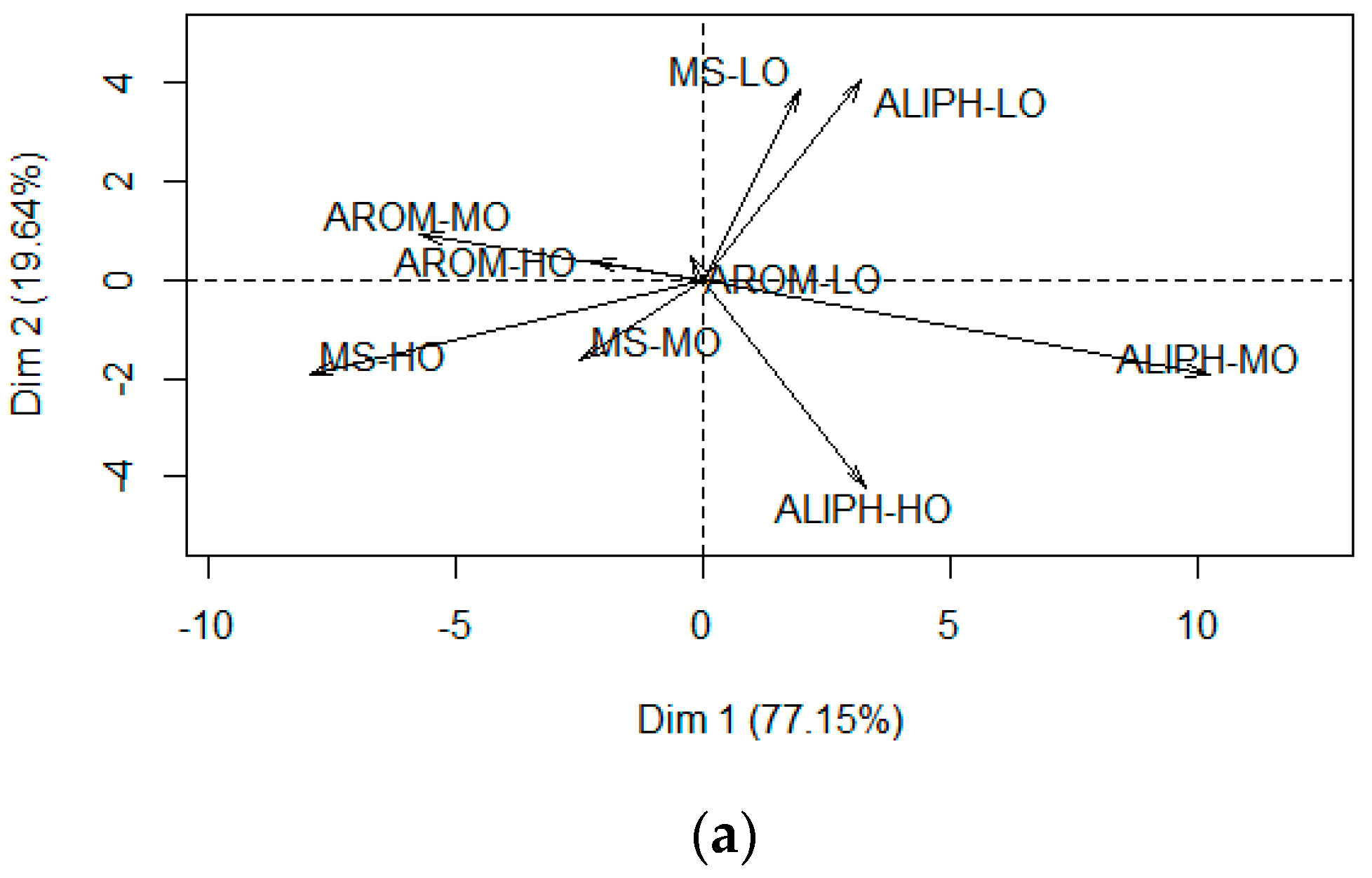
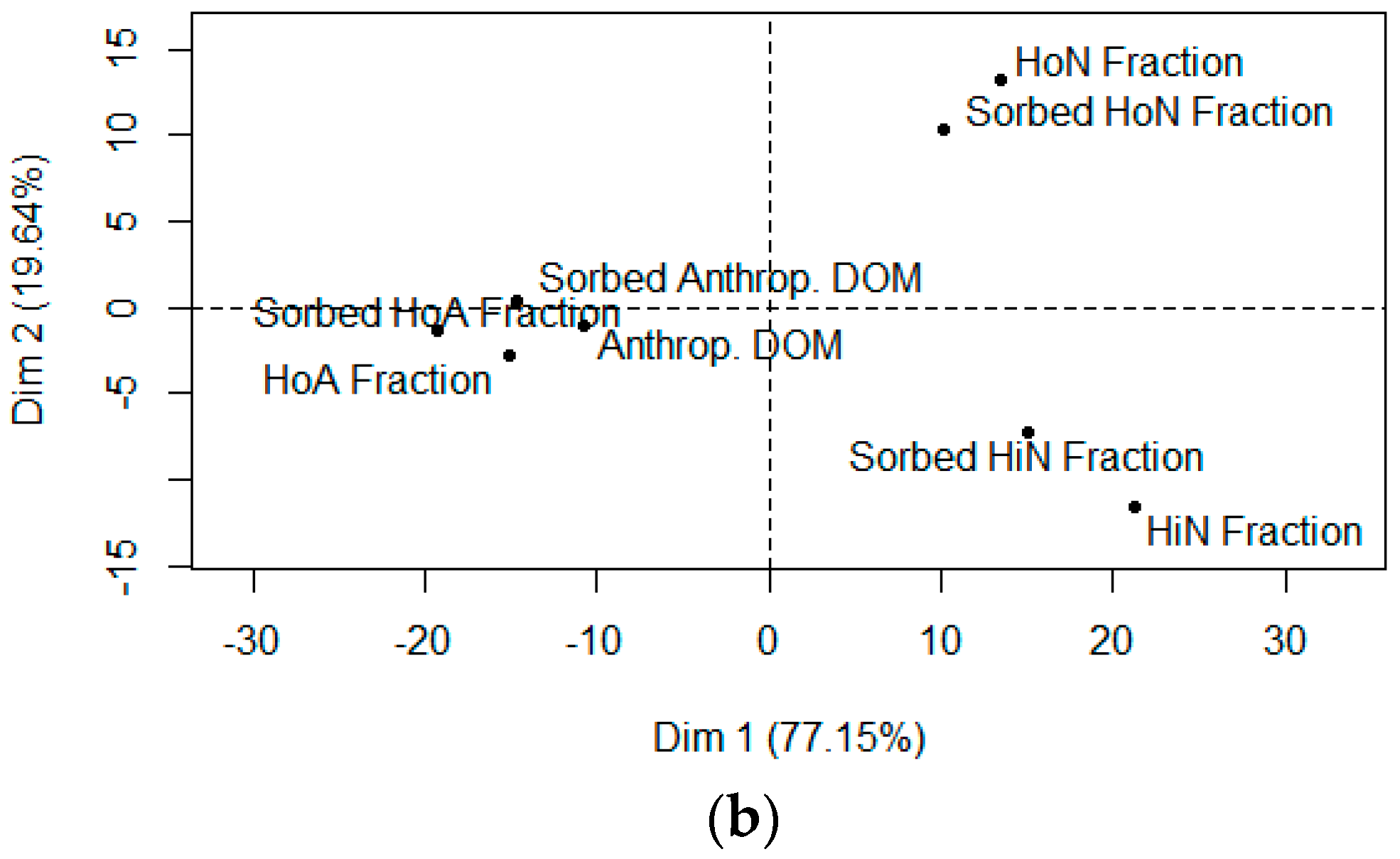
4.1. Composition-Dependent Sorption
4.2. Composition-Dependent Sorptive Fractionation
4.3. The Influence of Aromaticity, Oxygen Content, and Nitrogen Content on Sorptive Fractionation
4.4. The Influence of Non-Selective Sorption on the Formation of Mineral-Organic Associations
4.5. Additional Environmental Relevance
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Keiluweit, M.; Nico, P.S.; Kleber, M.; Fendorf, S. Are oxygen limitations under recognized regulators of organic carbon turnover in upland soils? Biogeochemistry 2016, 127, 157–171. [Google Scholar] [CrossRef]
- Boye, K.; Noël, V.; Tfaily, M.M.; Bone, S.E.; Williams, K.H.; Bargar, J.; Fendorf, S. Thermodynamically controlled preservation of organic carbon in floodplains. Nat. Geosci. 2017, 10, 415–419. [Google Scholar] [CrossRef]
- Young, I.M.; Crawford, J.W. Interactions and self-organization in the soil-microbe complex. Science 2004, 304, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.; Kleber, M. The contentious nature of soil organic matter. Nature 2015, 528, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L. Environmental Soil Chemistry; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Luthy, R.G.; Aiken, G.R.; Brusseau, M.L.; Cunningham, S.D.; Gschwend, P.M.; Pignatello, J.J.; Reinhard, M.; Traina, S.J.; Weber, W.J.; Westall, J.C. Sequestration of hydrophobic organic contaminants by geosorbents. Environ. Sci. Technol. 1997, 31, 3341–3347. [Google Scholar] [CrossRef]
- Miltner, A.; Bombach, P.; Schmidt-Brucken, B.; Kastner, M. SOM genesis: Microbial biomass as a significant source. Biogeochemistry 2012, 111, 41–55. [Google Scholar] [CrossRef]
- Kallenbach, C.M.; Frey, S.D.; Grandy, A.S. Direct evidence for microbial-derived soil organic matter formation and its ecophysiological controls. Nat. Commun. 2016, 7, 13630. [Google Scholar] [CrossRef] [PubMed]
- Hertkorn, N.; Frommberger, M.; Witt, M.; Koch, B.P.; Schmitt-Kopplin, P.; Perdue, E.M. Natural organic matter and the event horizon of mass spectrometry. Anal. Chem. 2008, 80, 8908–8919. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.; Kalbitz, K. Cycling downwards—Dissolved organic matter in soils. Soil Biol. Biochem. 2012, 52, 29–32. [Google Scholar] [CrossRef]
- Nebbioso, A.; Piccolo, A. Molecular characterization of dissolved organic matter (DOM): A critical review. Anal. Bioanal. Chem. 2013, 405, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Kogel-Knabner, I.; Guggenberger, G.; Kleber, M.; Kandeler, E.; Kalbitz, K.; Scheu, S.; Eusterhues, K.; Leinweber, P. Organo-mineral associations in temperate soils: Integrating biology, mineralogy, and organic matter chemistry. J. Plant Nutr. Soil Sci-Z. Pflanzenernahr. Bodenkund. 2008, 171, 61–82. [Google Scholar] [CrossRef]
- Kleber, M.; Eusterhues, K.; Keiluweit, M.; Mikutta, C.; Mikutta, R.; Nico, P.S. Mineral-organic associations: Formation, properties, and relevance in soil environments. In Advances in Agronomy; Sparks, D.L., Ed.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2015; Volume 130, pp. 1–140. [Google Scholar]
- Scott, E.E.; Rothstein, D.E. The dynamic exchange of dissolved organic matter percolating through six diverse soils. Soil Biol. Biochem. 2014, 69, 83–92. [Google Scholar] [CrossRef]
- Eusterhues, K.; Rennert, T.; Knicker, H.; Kogel-Knabner, I.; Totsche, K.U.; Schwertmann, U. Fractionation of organic matter due to reaction with ferrihydrite: Coprecipitation versus adsorption. Environ. Sci. Technol. 2011, 45, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Dynes, J.J.; Wang, J.; Sparks, D.L. Properties of Fe-organic matter associations via coprecipitation versus adsorption. Environ. Sci. Technol. 2014, 48, 13751–13759. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.; Sollins, P.; Sutton, R. A conceptual model of organo-mineral interactions in soils: Self-assembly of organic molecular fragments into zonal structures on mineral surfaces. Biogeochemistry 2007, 85, 9–24. [Google Scholar] [CrossRef]
- Solomon, D.; Lehmann, J.; Harden, J.; Wang, J.; Kinyangi, J.; Heymann, K.; Karunakaran, C.; Lu, Y.S.; Wirick, S.; Jacobsen, C. Micro- and nano-environments of carbon sequestration: Multi-element STXM-NEXAFS spectromicroscopy assessment of microbial carbon and mineral associations. Chem. Geol. 2012, 329, 53–73. [Google Scholar] [CrossRef]
- Chasse, A.W.; Ohno, T.; Higgins, S.R.; Amirbahman, A.; Yildirim, N.; Parr, T.B. Chemical force spectroscopy evidence supporting the layer-by-layer model of organic matter binding to iron (oxy)hydroxide mineral surfaces. Environ. Sci. Technol. 2015, 49, 9733–9741. [Google Scholar] [CrossRef] [PubMed]
- Masoom, H.; Courtier-Murias, D.; Farooq, H.; Soong, R.; Kelleher, B.P.; Zhang, C.; Maas, W.E.; Fey, M.; Kumar, R.; Monette, M.; et al. Soil organic matter in its native state: Unravelling the most complex biomaterial on Earth. Environ. Sci. Technol. 2016, 50, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Mueller, C.W.; Hoschen, C.; Buegger, F.; Heister, K.; Schulz, S.; Schloter, M.; Kogel-Knabner, I. Submicron structures provide preferential spots for carbon and nitrogen sequestration in soils. Nat. Commun. 2014, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Six, J.; Conant, R.T.; Paul, E.A.; Paustian, K. Stabilization mechanisms of soil organic matter: Implications for C-saturation of soils. Plant Soil 2002, 241, 155–176. [Google Scholar] [CrossRef]
- Guggenberger, G.; Kaiser, K. Dissolved organic matter in soil: Challenging the paradigm of sorptive preservation. Geoderma 2003, 113, 293–310. [Google Scholar] [CrossRef]
- Chorover, J.; Amistadi, M.K. Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces. Geochim. Cosmochim. Acta 2001, 65, 95–109. [Google Scholar] [CrossRef]
- Feng, X.J.; Simpson, A.J.; Simpson, M.J. Chemical and mineralogical controls on humic acid sorption to clay mineral surfaces. Org. Geochem. 2005, 36, 1553–1566. [Google Scholar] [CrossRef]
- Kramer, M.G.; Sanderman, J.; Chadwick, O.A.; Chorover, J.; Vitousek, P.M. Long-term carbon storage through retention of dissolved aromatic acids by reactive particles in soil. Glob. Chang. Biol. 2012, 18, 2594–2605. [Google Scholar] [CrossRef]
- Mitchell, P.J.; Simpson, A.J.; Soong, R.; Oren, A.; Chefetz, B.; Simpson, M.J. Solution-state NMR investigation of the sorptive fractionation of dissolved organic matter by alkaline mineral soils. Environ. Chem. 2013, 10, 333–340. [Google Scholar] [CrossRef]
- Fleury, G.; Del Nero, M.; Barillon, R. Effect of mineral surface properties (alumina, kaolinite) on the sorptive fractionation mechanisms of soil fulvic acids: Molecular-scale ESI-MS studies. Geochim. Cosmochim. Acta 2017, 196, 1–17. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, S.; Wang, S.; Luo, L.; Cao, D.; Christie, P. Molecular-scale investigation with ESI-FT-ICR-MS on fractionation of dissolved organic matter induced by adsorption on iron oxyhydroxides. Environ. Sci. Technol. 2016, 50, 2328–2336. [Google Scholar] [CrossRef] [PubMed]
- Avneri-Katz, S.; Young, R.B.; Mckenna, A.M.; Chen, H.; Corilo, Y.E.; Polubesova, T.; Borch, T.; Chefetz, B. Adsorptive fractionation of dissolved organic matter (DOM) by mineral soil: Macroscale approach and molecular insight. Org. Geochem. 2017, 103, 113–124. [Google Scholar] [CrossRef]
- Minor, E.C.; Steinbring, C.J.; Longnecker, K.; Kujawinski, E.B. Characterization of dissolved organic matter in Lake Superior and its watershed using ultrahigh resolution mass spectrometry. Org. Geochem. 2012, 43, 1–11. [Google Scholar] [CrossRef]
- Chefetz, B.; Hatcher, P.G.; Hadar, Y.; Chen, Y.N. Characterization of dissolved organic matter extracted from composted municipal solid waste. Soil Sci. Soc. Am. J. 1998, 62, 326–332. [Google Scholar] [CrossRef]
- Chefetz, B.; Illani, T.; Schulz, E.; Chorover, J. Wastewater dissolved organic matter: Characteristics and sorptive capabilities. Water Sci. Technol. 2006, 53, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Von Lutzow, M.; Kogel-Knabner, I.; Ekschmitt, K.; Matzner, E.; Guggenberger, G.; Marschner, B.; Flessa, H. Stabilization of organic matter in temperate soils: Mechanisms and their relevance under different soil conditions—A review. Eur. J. Soil Sci. 2006, 57, 426–445. [Google Scholar] [CrossRef]
- Polubesova, T.; Chen, Y.; Navon, R.; Chefetz, B. Interactions of hydrophobic fractions of dissolved organic matter with Fe(3+)- and Cu(2+)-montmorillonite. Environ. Sci. Technol. 2008, 42, 4797–4803. [Google Scholar] [CrossRef] [PubMed]
- Olshansky, Y.; Polubesova, T.; Chefetz, B. Reconstitution of cutin monomers on smectite surfaces: Adsorption and esterification. Geoderma 2014, 232, 406–413. [Google Scholar] [CrossRef]
- Schwertmann, U. Solubility and dissolution of iron oxides. Plant Soil 1991, 130, 1–25. [Google Scholar] [CrossRef]
- Stefánsson, A. Iron(III) hydrolysis and solubility at 25 °C. Environ. Sci. Technol. 2007, 41, 6117–6123. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.M.L.; Bertoncini, E.I.; Abreu, C.H. Composting sewage sludge with green waste from tree pruning. Sci. Agricola 2015, 72, 432–439. [Google Scholar] [CrossRef]
- Yanez, R.; Alonso, J.L.; Diaz, M.J. Influence of bulking agent on sewage sludge composting process. Bioresour. Technol. 2009, 100, 5827–5833. [Google Scholar] [CrossRef] [PubMed]
- Amery, F.; Vanmoorleghem, C.; Smolders, E. Adapted DAX-8 fractionation method for dissolved organic matter (DOM) from soils: Development, calibration with test components and application to contrasting soil solutions. Eur. J. Soil Sci. 2009, 60, 956–965. [Google Scholar] [CrossRef]
- Dittmar, T.; Koch, B.; Hertkorn, N.; Kattner, G. A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater. Limnol. Oceanogr.-Meth. 2008, 6, 230–235. [Google Scholar] [CrossRef]
- Li, Y.; Harir, M.; Lucio, M.; Kanawati, B.; Smirnov, K.; Flerus, R.; Koch, B.P.; Schmitt-Kopplin, P.; Hertkorn, N. Proposed guidelines for solid phase extraction of Suwannee River dissolved organic matter. Anal. Chem. 2016, 88, 6680–6688. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, N.K.; Quinn, J.P.; Blakney, G.T.; Hendrickson, C.L.; Marshall, A.G. A novel 9.4 Tesla FTICR mass spectrometer with improved sensitivity, mass resolution, and mass range. J. Am. Soc. Mass Spectrom. 2011, 22, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Blakney, G.T.; Hendrickson, C.L.; Marshall, A.G. Predator data station: A fast data acquisition system for advanced FT-ICR MS experiments. Int. J. Mass Spectrom. 2011, 306, 246–252. [Google Scholar] [CrossRef]
- Senko, M.W.; Hendrickson, C.L.; Emmett, M.R.; Shi, S.D.H.; Marshall, A.G. External accumulation of ions for enhanced electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 1997, 8, 970–976. [Google Scholar] [CrossRef]
- Kaiser, N.K.; Savory, J.J.; Mckenna, A.M.; Quinn, J.P.; Hendrickson, C.L.; Marshall, A.G. Electrically compensated Fourier transform ion cyclotron resonance cell for complex mixture mass analysis. Anal. Chem. 2011, 83, 6907–6910. [Google Scholar] [CrossRef] [PubMed]
- Tolmachev, A.V.; Robinson, E.W.; Wu, S.; Kang, H.; Lourette, N.M.; Pasa-Tolic, L.; Smith, R.D. Trapped-ion cell with improved DC potential harmonicity for FT-ICR MS. J. Am. Soc. Mass Spectrom. 2008, 19, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Xian, F.; Hendrickson, C.L.; Blakney, G.T.; Beu, S.C.; Marshall, A.G. Automated broadband phase correction of Fourier transform ion cyclotron resonance mass spectra. Anal. Chem. 2010, 82, 8807–8812. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.D.H.; Drader, J.J.; Freitas, M.A.; Hendrickson, C.L.; Marshall, A.G. Comparison and interconversion of the two most common frequency-to-mass calibration functions for Fourier transform ion cyclotron resonance mass spectrometry2. Int. J. Mass Spectrom. 2000, 195–196, 591–598. [Google Scholar] [CrossRef]
- Ledford, E.B.; Rempel, D.L.; Gross, M.L. Space charge effects in Fourier transform mass spectrometry—MASS calibration. Anal. Chem. 1984, 56, 2744–2748. [Google Scholar] [CrossRef] [PubMed]
- Savory, J.J.; Kaiser, N.K.; Mckenna, A.M.; Xian, F.; Blakney, G.T.; Rodgers, R.P.; Hendrickson, C.L.; Marshall, A.G. Parts-per-billion Fourier transform ion cyclotron resonance mass measurement accuracy with a “walking” calibration equation. Anal. Chem. 2011, 83, 1732–1736. [Google Scholar] [CrossRef] [PubMed]
- Corilo, Y.E. PetroOrg; The Florida State University: Tallahassee, FL, USA, 2012. [Google Scholar]
- Kendrick, E. A mass scale based on CH2 = 14.0000 for high resolution mass spectrometry of organic compounds. Anal. Chem. 1963, 35. [Google Scholar] [CrossRef]
- Hughey, C.A.; Hendrickson, C.L.; Rodgers, R.P.; Marshall, A.G.; Qian, K.N. Kendrick mass defect spectrum: A compact visual analysis for ultrahigh-resolution broadband mass spectra. Anal. Chem. 2001, 73, 4676–4681. [Google Scholar] [CrossRef] [PubMed]
- Koch, B.P.; Dittmar, T. From mass to structure: An aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun. Mass Spectrom. 2006, 20, 926–932. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Le, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- D’andrilli, J.; Cooper, W.T.; Foreman, C.M.; Marshall, A.G. An ultrahigh-resolution mass spectrometry index to estimate natural organic matter lability. Rapid Commun. Mass Spectrom. 2015, 29, 2385–2401. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, N.; Dairpoosh, F.; Yassin, G.; Golon, A.; Jaiswal, R. What is under the hump? Mass spectrometry based analysis of complex mixtures in processed food—Lessons from the characterisation of black tea thearubigins, coffee melanoidines and caramel. Food Funct. 2013, 4, 1130–1147. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, D.C.; Chen, H.M.; Willoughby, A.S.; Hatcher, P.G. Formation of black carbon-like and alicyclic aliphatic compounds by hydroxyl radical initiated degradation of lignin. Org. Geochem. 2015, 82, 69–76. [Google Scholar] [CrossRef]
- Hertkorn, N.; Ruecker, C.; Meringer, M.; Gugisch, R.; Frommberger, M.; Perdue, E.M.; Witt, M.; Schmitt-Kopplin, P. High-precision frequency measurements: Indispensable tools at the core of the molecular-level analysis of complex systems. Anal. Bioanal. Chem. 2007, 389, 1311–1327. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Frandsen, C.; Wallace, A.F.; Legg, B.; Khalid, S.; Zhang, H.; Mørup, S.; Banfield, J.F.; Waychunas, G.A. Precipitation pathways for ferrihydrite formation in acidic solutions. Geochim. Cosmochim. Acta 2016, 172, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Gu, B.H.; Schmitt, J.; Chen, Z.H.; Liang, L.Y.; Mccarthy, J.F. Adsorption and desorption of natural organic matter on iron oxide—Mechanisms and models. Environ. Sci. Technol. 1994, 28, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Brunner, P.H.; Capri, S.; Marcomini, A.; Giger, W. Occurrence and behavior of linear alkulbenzenesulfonates, nonylphenol, nonylphenol monophenol and nonylphenol diethoxylates in sewage and sewage sludge treatment. Water Res. 1988, 22, 1465–1472. [Google Scholar] [CrossRef]
- Gonsior, M.; Zwartjes, M.; Cooper, W.J.; Song, W.; Ishida, K.P.; Tseng, L.Y.; Jeung, M.K.; Rosso, D.; Hertkorn, N.; Schmitt-Kopplin, P. Molecular characterization of effluent organic matter identified by ultrahigh resolution mass spectrometry. Water Res. 2011, 45, 2943–2953. [Google Scholar] [CrossRef] [PubMed]
- Cantarero, S.; Prieto, C.A.; Lopez, I. Occurrence of high-tonnage anionic surfactants in Spanish sewage sludge. J. Environ. Manag. 2012, 95, S149–S153. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Singer, H.P.; Longree, P.; Loos, M.; Ruff, M.; Stravs, M.A.; Vidal, C.R.; Hollender, J. Strategies to characterize polar organic contamination in wastewater: Exploring the capability of high resolution mass spectrometry. Environ. Sci. Technol. 2014, 48, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Knorr, K.H. DOC-dynamics in a small headwater catchment as driven by redox fluctuations and hydrological flow paths—Are DOC exports mediated by iron reduction/oxidation cycles? Biogeosciences 2013, 10, 891–904. [Google Scholar] [CrossRef]
- Tombacz, E.; Libor, Z.; Illes, E.; Majzik, A.; Klumpp, E. The role of reactive surface sites and complexation by humic acids in the interaction of clay mineral and iron oxide particles. Org. Geochem. 2004, 35, 257–267. [Google Scholar] [CrossRef]
- Arnarson, T.S.; Keil, R.G. Mechanisms of pore water organic matter adsorption to montmorillonite. Mar. Chem. 2000, 71, 309–320. [Google Scholar] [CrossRef]
- Leenheer, J.A.; Wershaw, R.L.; Reddy, M.M. Strong-acid, carboxyl group structures in fulvic acid from the Suwannee River, Georgia. 1. Minor structures. Environ. Sci. Technol. 1995, 29, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Leenheer, J.A.; Wershaw, R.L.; Reddy, M.M. Strong-acid, carboxyl group structures in fulvic acid from the Suwannee River, Georgia. 2. Major structures. Environ. Sci. Technol. 1995, 29, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Bingham, A.H.; Cotrufo, M.F. Organic nitrogen storage in mineral soil: Implications for policy and management. Sci. Total Environ. 2016, 551–552, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Genest, S.C.; Simpson, M.J.; Simpson, A.J.; Soong, R.; Mcnally, D.J. Analysis of soil organic matter at the solid-water interface by nuclear magnetic resonance spectroscopy. Environ. Chem. 2014, 11, 472–482. [Google Scholar] [CrossRef]
- Kinney, C.A.; Furlong, E.T.; Zaugg, S.D.; Burkhardt, M.R.; Werner, S.L.; Cahill, J.D.; Jorgensen, G.R. Survey of organic wastewater contaminants in biosolids destined for land application. Environ. Sci. Technol. 2006, 40, 7207–7215. [Google Scholar] [CrossRef] [PubMed]
- Cogger, C.G.; Forge, T.A.; Neilsen, G.H. Biosolids recycling: Nitrogen management and soil ecology. Can. J. Soil Sci. 2006, 86, 613–620. [Google Scholar] [CrossRef]
- Gray, J.L.; Borch, T.; Furlong, E.T.; Davis, J.G.; Yager, T.J.; Yang, Y.Y.; Kolpin, D.W. Rainfall-runoff of anthropogenic waste indicators from agricultural fields applied with municipal biosolids. Sci. Total Environ. 2017, 580, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Zbytniewski, R.; Buszewski, B. Characterization of natural organic matter (NOM) derived from sewage sludge compost. Part 1: Chemical and spectroscopic properties. Bioresour. Technol. 2005, 96, 471–478. [Google Scholar] [CrossRef] [PubMed]
- D’andrilli, J.; Foreman, C.M.; Marshall, A.G.; Mcknight, D.M. Characterization of IHSS Pony Lake fulvic acid dissolved organic matter by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry and fluorescence spectroscopy. Org. Geochem. 2013, 65, 19–28. [Google Scholar] [CrossRef]
- Jardé, E.; Mansuy, L.; Faure, P. Organic markers in the lipidic fraction of sewage sludges. Water Res. 2005, 39, 1215–1232. [Google Scholar] [CrossRef] [PubMed]
- Ljung, K.; Maley, F.; Cook, A.; Weinstein, P. Acid sulfate soils and human health—A Millennium Ecosystem Assessment. Environ. Int. 2009, 35, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Jayalath, N.; Mosley, L.M.; Fitzpatrick, R.W.; Marschner, P. Addition of organic matter influences pH changes in reduced and oxidised acid sulfate soils. Geoderma 2016, 262, 125–132. [Google Scholar] [CrossRef]
- Kolbl, A.; Marschner, P.; Fitzpatrick, R.; Mosley, L.; Kogel-Knabner, I. Linking organic matter composition in acid sulfate soils to pH recovery after re-submerging. Geoderma 2017, 308, 350–362. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | Initial DOC (mg C/L) | Sorbed DOC (mg C/g dry clay) | Sorbed DOC (%) |
|---|---|---|---|
| Anthropogenic DOM | 54 ± 1 | 9.8 ± 0.4 | 54 ± 2 |
| HoA Fraction | 55 ± 0.9 | 11.8 ± 0.5 | 64 ± 3 |
| HoN Fraction | 39 | 7.6 | 58 |
| HiN Fraction | 14 ± 2 | 2.1 ± 0.2 | 45 ± 4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, R.B.; Avneri-Katz, S.; McKenna, A.M.; Chen, H.; Bahureksa, W.; Polubesova, T.; Chefetz, B.; Borch, T. Composition-Dependent Sorptive Fractionation of Anthropogenic Dissolved Organic Matter by Fe(III)-Montmorillonite. Soil Syst. 2018, 2, 14. https://doi.org/10.3390/soilsystems2010014
Young RB, Avneri-Katz S, McKenna AM, Chen H, Bahureksa W, Polubesova T, Chefetz B, Borch T. Composition-Dependent Sorptive Fractionation of Anthropogenic Dissolved Organic Matter by Fe(III)-Montmorillonite. Soil Systems. 2018; 2(1):14. https://doi.org/10.3390/soilsystems2010014
Chicago/Turabian StyleYoung, Robert B., Shani Avneri-Katz, Amy M. McKenna, Huan Chen, William Bahureksa, Tamara Polubesova, Benny Chefetz, and Thomas Borch. 2018. "Composition-Dependent Sorptive Fractionation of Anthropogenic Dissolved Organic Matter by Fe(III)-Montmorillonite" Soil Systems 2, no. 1: 14. https://doi.org/10.3390/soilsystems2010014
APA StyleYoung, R. B., Avneri-Katz, S., McKenna, A. M., Chen, H., Bahureksa, W., Polubesova, T., Chefetz, B., & Borch, T. (2018). Composition-Dependent Sorptive Fractionation of Anthropogenic Dissolved Organic Matter by Fe(III)-Montmorillonite. Soil Systems, 2(1), 14. https://doi.org/10.3390/soilsystems2010014