Speciation and Mobility of Mercury in Soils Contaminated by Legacy Emissions from a Chemical Factory in the Rhône Valley in Canton of Valais, Switzerland

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area and Contamination History
2.2. Soil Sampling
2.2.1. Soil Samples Following VBBo Procedures
2.2.2. Soil Samples Collected as Undisturbed Cores
2.2.3. Sediment Samples from the Factory Site
2.3. Soil Analyses
2.4. Total Hg Analyses
2.5. Hg Speciation
2.5.1. Sequential Extractions of Hg
2.5.2. Thermal Desorption Analysis of Hg
2.5.3. Methyl-Hg Analysis
2.6. Hg Release Potential
2.6.1. Influence of CaCl2
2.6.2. Influence of Redox Processes on Hg Release
3. Results
3.1. Sample Characterization and Total Hg Determination
3.1.1. Total Hg in Soils and Sediments
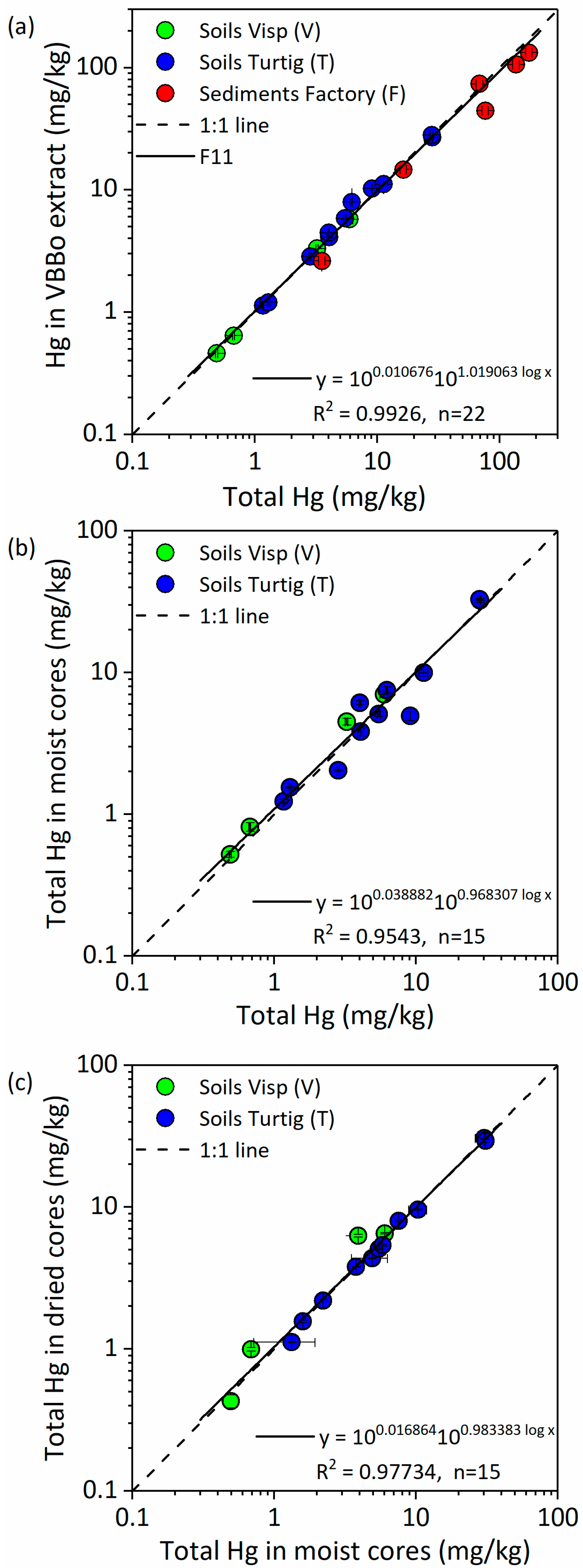
3.1.2. Effect of Soil Sampling Method
3.1.3. Effect of Sample Drying
3.2. Hg Speciation
3.2.1. Hg Pools Determined by Sequential Extraction
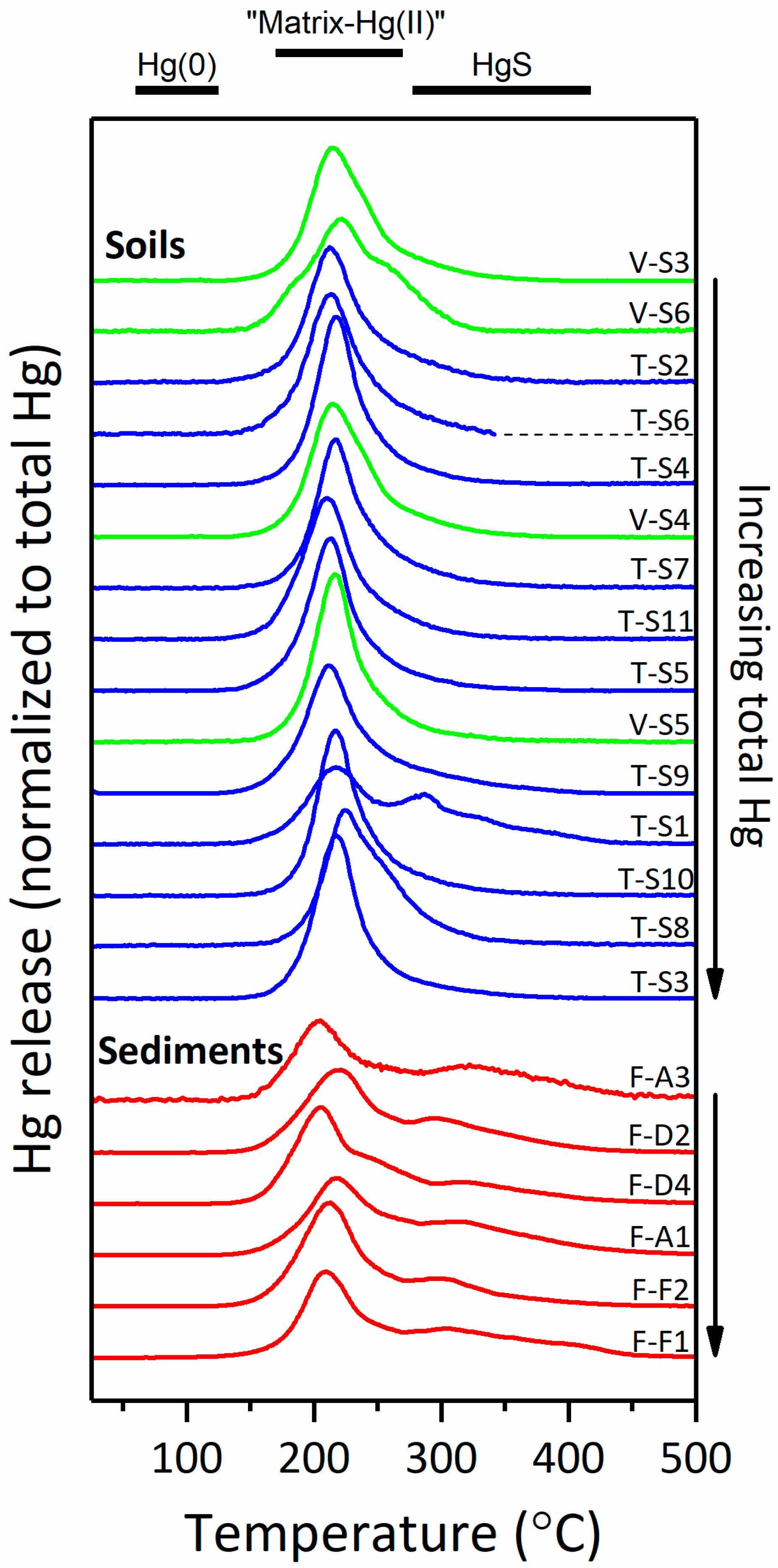
3.2.2. Thermal Desorption Analysis of Hg
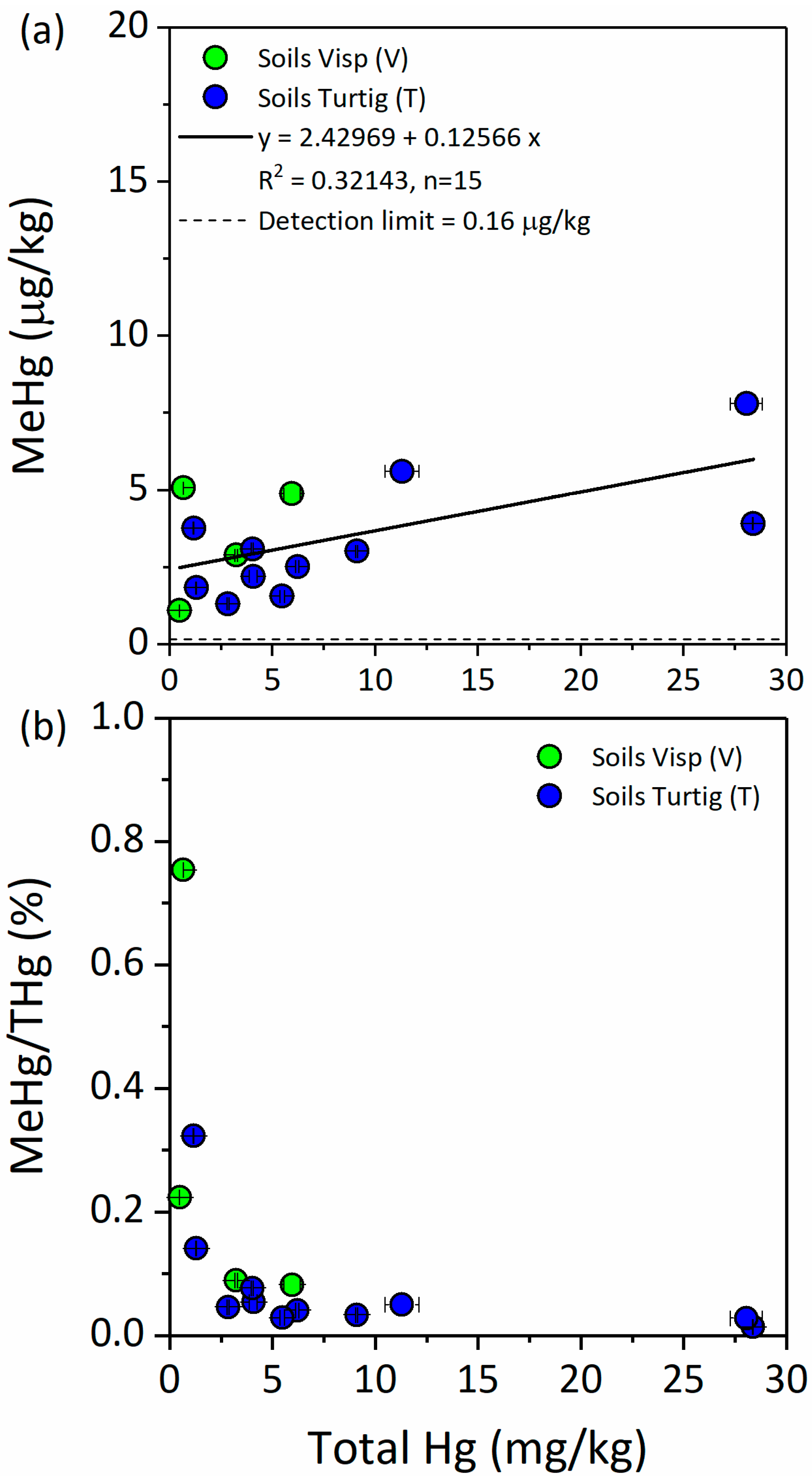
3.2.3. Methyl-Hg
3.3. Hg Release Potential from Soils and Sediments
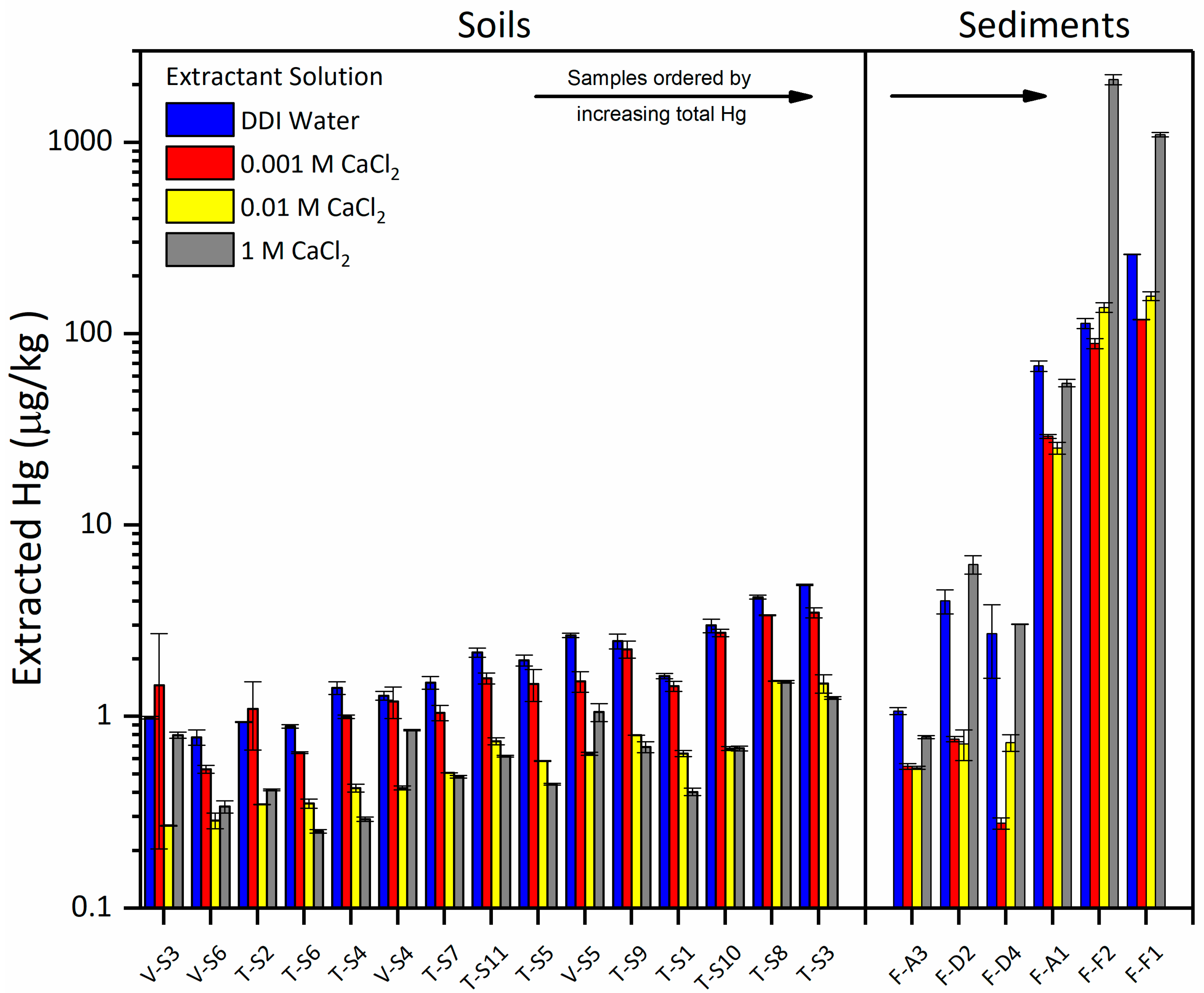
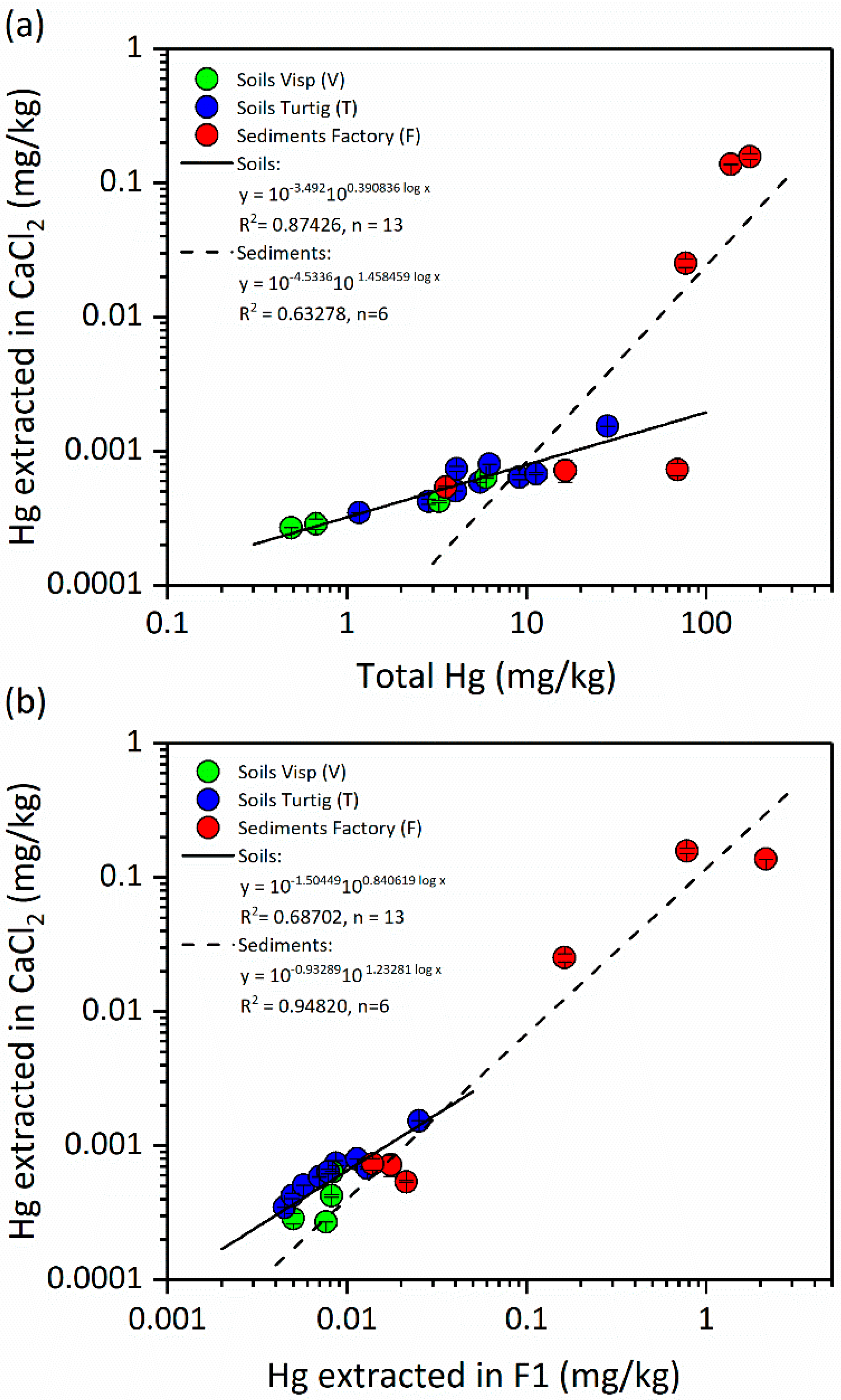
3.3.1. Effect of CaCl2 on Hg Solubility
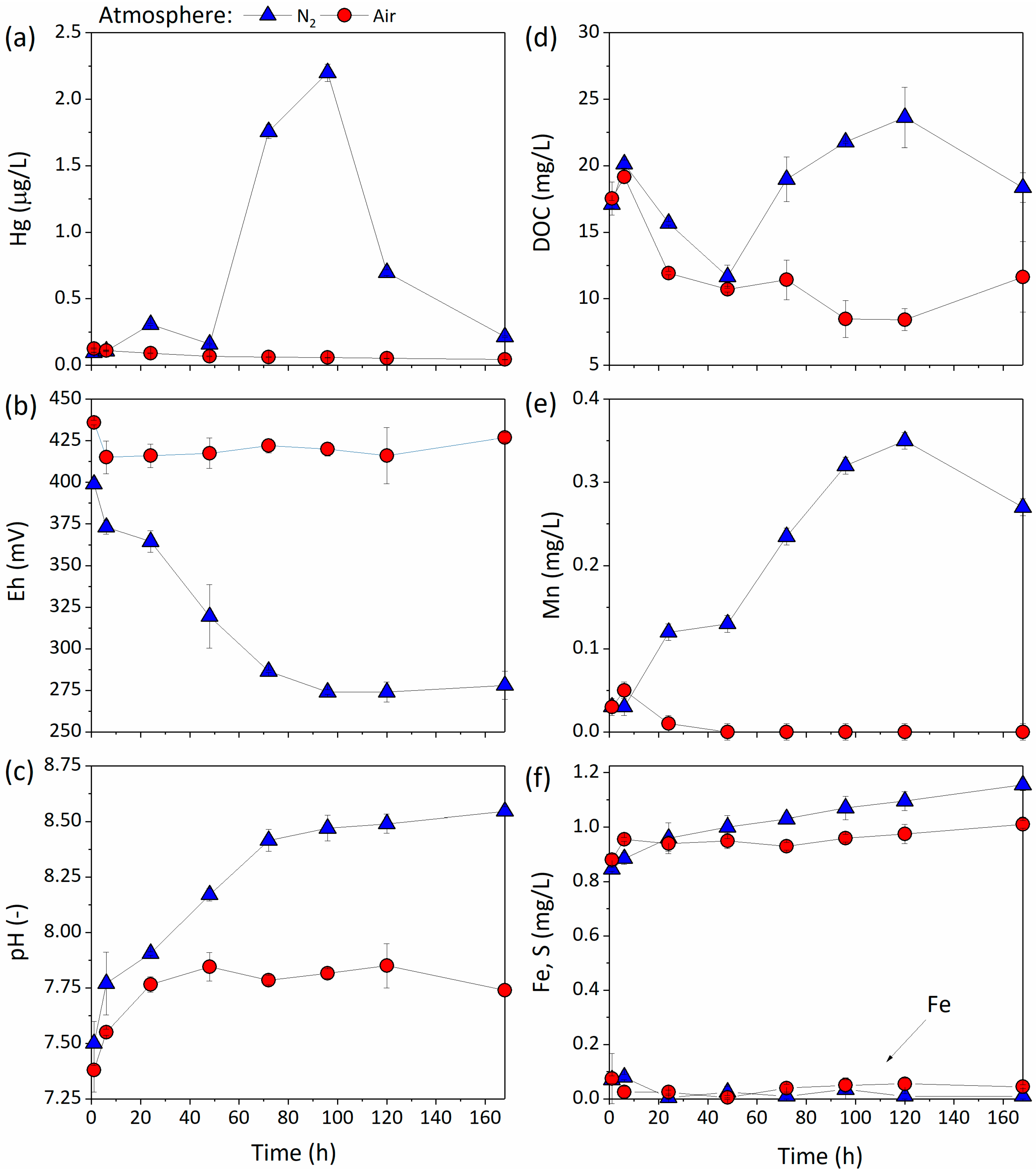
3.3.2. Effect of Redox Processes on Hg Mobility
4. Discussion
4.1. Total Hg in Soils and Sediments
4.2. Hg Species in Soils and Factory Sediments
4.3. Mobility of Hg
4.4. Environmental Implications
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Selin, N.E. Global biogeochemical cycling of mercury: A review. Annu. Rev. Environ. Resour. 2009, 34, 43–63. [Google Scholar] [CrossRef]
- Mason, R.P.; Choi, A.L.; Fitzgerald, W.F.; Hammerschmidt, C.R.; Lamborg, C.H.; Soerensen, A.L.; Sunderland, E.M. Mercury biogeochemical cycling in the ocean and policy implications. Environ. Res. 2012, 119, 101–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, R.P.; Fitzgerald, W.F.; Morel, F.M.M. The biogeochemical cycling of elemental mercury—Anthropogenic influences. Geochim. Cosmochim. Acta 1994, 58, 3191–3198. [Google Scholar] [CrossRef]
- Fitzgerald, W.F.; Lamborg, C.H. Geochemistry of Mercury in the Environment. In Treatise on Geochemistry, 2nd ed.; Holland, H.D., Turekian, K.K., Eds.; Elsevier: Oxford, UK, 2014; pp. 91–129. [Google Scholar]
- Obrist, D.; Pokharel, A.K.; Moore, C. Vertical profile measurements of soil air suggest immobilization of gaseous elemental mercury in mineral soil. Environ. Sci. Technol. 2014, 48, 2242–2252. [Google Scholar] [CrossRef] [PubMed]
- Schwab, P.; Dahinden, R.; Desaules, A. Einflüsse der Probentrocknung auf Quecksilberkonzentrationen in Bodenproben. Bull. BGS 2002, 26, 39–42. [Google Scholar]
- Gabriel, M.C.; Williamson, D.G. Principal biogeochemical factors affecting the speciation and transport of mercury through the terrestrial environment. Environ. Geochem. Health 2004, 26, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Schuster, E. The behavior of mercury in the soil with special emphasis on complexation and adsorption processes—A review of the literature. Water Air Soil Pollut. 1991, 56, 667–680. [Google Scholar] [CrossRef]
- Skyllberg, U. Chemical Speciation of Mercury in Soil and Sediment. In Environmental Chemistry and Toxicology of Mercury; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 219–258. [Google Scholar]
- Poulin, B.A.; Aiken, G.R.; Nagy, K.L.; Manceau, A.; Krabbenhoft, D.P.; Ryan, J.N. Mercury transformation and release differs with depth and time in a contaminated riparian soil during simulated flooding. Geochim. Cosmochim. Acta 2016, 176, 118–138. [Google Scholar] [CrossRef] [Green Version]
- Wallschlager, D.; Desai, M.V.M.; Spengler, M.; Windmoller, C.C.; Wilken, R.D. How humic substances dominate mercury geochemistry in contaminated floodplain soils and sediments. J. Environ. Qual. 1998, 27, 1044–1054. [Google Scholar] [CrossRef]
- Wallschlager, D.; Desai, M.V.M.; Wilken, R.D. The role of humic substances in the aqueous mobilization of mercury from contaminated floodplain soils. Water Air Soil Pollut. 1996, 90, 507–520. [Google Scholar] [CrossRef]
- Yin, Y.J.; Allen, H.E.; Huang, C.P.; Sparks, D.L.; Sanders, P.F. Kinetics of mercury(II) adsorption and desorption on soil. Environ. Sci. Technol. 1997, 31, 496–503. [Google Scholar] [CrossRef]
- Kim, C.S.; Rytuba, J.; Brown, G.E., Jr. EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides—II. Effects of chloride and sulfate. J. Colloid Interface Sci. 2004, 270, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Charlet, L.; Blancho, F.; Bonnet, T.; Garambois, S.; Boivin, P.; Ferber, T.; Tisserand, D.; Guedron, S. Industrial mercury pollution in a mountain valley: A combined geophysical and geochemical study. Procedia Earth Planet. Sci. 2017, 17, 77–80. [Google Scholar] [CrossRef]
- Ravichandran, M.; Aiken, G.R.; Ryan, J.N.; Reddy, M.M. Inhibition of precipitation and aggregation of metacinnabar (mercuric sulfide) by dissolved organic matter isolated from the Florida Everglades. Environ. Sci. Technol. 1999, 33, 1418–1423. [Google Scholar] [CrossRef]
- Gerbig, C.A.; Kim, C.S.; Stegemeier, J.P.; Ryan, J.N.; Aiken, G.R. Formation of nanocolloidal metacinnabar in mercury-DOM-sulfide systems. Environ. Sci. Technol. 2011, 45, 9180–9187. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.O.; Harris, L.A.; Turner, R.R.; Henson, T.J.; Melton, R.E.; Stevenson, R.J. Characterization of mercury species in contaminated floodplain soils. Water Air Soil Pollut. 1995, 80, 1105–1108. [Google Scholar] [CrossRef]
- Barnett, M.O.; Harris, L.A.; Turner, R.R.; Stevenson, R.J.; Henson, T.J.; Melton, R.C.; Hoffman, D.P. Formation of mercuric sulfide in soil. Environ. Sci. Technol. 1997, 31, 3037–3043. [Google Scholar] [CrossRef]
- Drott, A.; Bjorn, E.; Bouchet, S.; Skyllberg, U. Refining thermodynamic constants for mercury(II)-sulfides in equilibrium with metacinnabar at sub-micromolar aqueous sulfide concentrations. Environ. Sci. Technol. 2013, 47, 4197–4203. [Google Scholar] [CrossRef] [PubMed]
- Deonarine, A.; Hsu-Kim, H. Precipitation of mercuric sulfide nanoparticles in NOM-containing water: Implications for the natural environment. Environ. Sci. Technol. 2009, 43, 2368–2373. [Google Scholar] [CrossRef] [PubMed]
- Poulin, B.A.; Gerbig, C.A.; Kim, C.S.; Stegemeier, J.P.; Ryan, J.N.; Aiken, G.R. Effects of sulfide concentration and dissolved organic matter characteristics on the structure of nanocolloidal metacinnabar. Environ. Sci. Technol. 2017, 51, 13133–13142. [Google Scholar] [CrossRef] [PubMed]
- Graham, A.M.; Aiken, G.R.; Gilmour, C.C. Dissolved organic matter enhances microbial mercury methylation under sulfidic conditions. Environ. Sci. Technol. 2012, 46, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kim, B.; Leyard, C.; Reinsch, B.C.; Lowry, G.V.; Deshusses, M.A.; Hsu-Kim, H. Methylation of mercury by bacteria exposed to dissolved, nanoparticulate, and microparticulate mercuric sulfides. Environ. Sci. Technol. 2012, 46, 6950–6958. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, T.W.; Magos, L. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 2006, 36, 609–662. [Google Scholar] [CrossRef] [PubMed]
- Barkay, T.; Wagner-Dobler, I. Microbial transformations of mercury: Potentials, challenges, and achievements in controlling mercury toxicity in the environment. Adv. Appl. Microbiol. 2005, 57, 1–52. [Google Scholar] [PubMed]
- Bloom, N.S.; Preus, E.; Katon, J.; Hiltner, M. Selective extractions to assess the biogeochemically relevant fractionation of inorganic mercury in sediments and soils. Anal. Chim. Acta 2003, 479, 233–248. [Google Scholar] [CrossRef]
- Issaro, N.; Abi-Ghanem, C.; Bermond, A. Fractionation studies of mercury in soils and sediments: A review of the chemical reagents used for mercury extraction. Anal. Chim. Acta 2009, 631, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nirel, P.M.V.; Morel, F.M.M. Pitfalls of Sequential Extractions. Water Res. 1990, 24, 1055–1056. [Google Scholar] [CrossRef]
- Reis, A.T.; Davidson, C.M.; Vale, C.; Pereira, E. Overview and challenges of mercury fractionation and speciation in soils. Trac-Trends Anal. Chem. 2016, 82, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Biester, H.; Scholz, C. Determination of mercury binding forms in contaminated soils: Mercury pyrolysis versus sequential extractions. Environ. Sci. Technol. 1997, 31, 233–239. [Google Scholar] [CrossRef]
- Reis, A.T.; Coelho, J.P.; Rucandio, I.; Davidson, C.M.; Duarte, A.C.; Pereira, E. Thermo-desorption: A valid tool for mercury speciation in soils and sediments? Geoderma 2015, 237, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Das Bundesamt für Umwelt (BAFU). Vollzugshilfe: Erläuterungen zur Verordnung vom 1. Juli 1998 über Belastungen des Bodens (VBBo); BAFU: Bern, Switzerland, 2001. [Google Scholar]
- Christian, G.; Jean-Rober, E. Voruntersuchung von Belasteten Standorten—Historische Untersuchung: Objekt Grossgrundkanal; ForumUmwelt AG: Visp, Switzerland, 2011. [Google Scholar]
- VBBo. Verordnung über Belastungen des Bodens; VBBo: Bern, Switzerland, 1998. [Google Scholar]
- AltIV (Altlasten-Verordnung). Verordnung über die Sanierung von belasteten Standorten; AltIV: Bern, Switzerland, 1998. [Google Scholar]
- Biester, H.; Nehrke, G. Quantification of mercury in soils and sediments—Acid digestion versus pyrolysis. Fresen. J. Anal. Chem. 1997, 358, 446–452. [Google Scholar]
- VVEA (Abfallverordnung). Verordnung über die Vermeidung und die Entsorgung von Abfällen; VVEA: Bern, Switzerland, 2016. [Google Scholar]
- Kim, C.S.; Bloom, N.S.; Rytuba, J.J.; Brown, G.E., Jr. Mercury speciation by X-ray absorption fine structure spectroscopy and sequential chemical extractions: A comparison of speciation methods. Environ. Sci. Technol. 2003, 37, 5102–5108. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, D.; Guo, H.B.; Parks, J.M.; Gu, B.H.; Summers, A.O.; Miller, S.M.; Liang, L.Y.; Smith, J.C. Why mercury prefers soft ligands. J. Phys. Chem. Lett. 2013, 4, 2317–2322. [Google Scholar] [CrossRef]
- Skyllberg, U. Competition among thiols and inorganic sulfides and polysulfides for Hg and MeHg in wetland soils and sediments under suboxic conditions: Illumination of controversies and implications for MeHg net production. J. Geophys. Res. 2008, 113, G00C03. [Google Scholar] [CrossRef]
- Wallschlager, D.; Desai, M.V.M.; Spengler, M.; Wilken, R.D. Mercury speciation in floodplain soils and sediments along a contaminated river transect. J. Environ. Qual. 1998, 27, 1034–1044. [Google Scholar] [CrossRef]
- Skyllberg, U.; Qian, J.; Frech, W.; Xia, K.; Bleam, W.F. Distribution of mercury, methyl mercury and organic sulphur species in soil, soil solution and stream of a boreal forest catchment. Biogeochemistry 2003, 64, 53–76. [Google Scholar] [CrossRef]
- Skyllberg, U.; Bloom, P.R.; Qian, J.; Lin, C.M.; Bleam, W.F. Complexation of mercury (II) in soil organic matter: EXAFS evidence for linear two-coordination with reduced sulfur groups. Environ. Sci. Technol. 2006, 40, 4174–4180. [Google Scholar] [CrossRef] [PubMed]
- Grigg, A.R.C.; Kretzschmar, R.; Gilli, R.S.; Wiederhold, J.G. Mercury isotope signatures of digests and sequential extractions from industrially contaminated soils and sediments. Sci. Total Environ. 2018, 636, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.E.; Hines, M.E.; Higueras, P.L.; Adatto, I.; Lasorsa, B.K. Mercury speciation and microbial transformations in mine wastes, stream sediments, and surface waters at the Almaden Mining District, Spain. Environ. Sci. Technol. 2004, 38, 4285–4292. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.S.; Wiederhold, J.G.; Jew, A.D.; Brown, G.E., Jr.; Bourdon, B.; Kretzschmar, R. Small-scale studies of roasted ore waste reveal extreme ranges of stable mercury isotope signatures. Geochim. Cosmochim. Acta 2014, 137, 1–17. [Google Scholar] [CrossRef]
- Hall, G.E.M.; Pelchat, P.; Percival, J.B. The design and application of sequential extractions for mercury, Part 1. Optimization of HNO3 extraction for all non-sulphide forms of Hg. Geochem. Explor. Environ. Anal. 2005, 5, 107–113. [Google Scholar] [CrossRef]
- Tauson, V.L.; Akimov, V.V. Introduction to the theory of forced equilibria: General principles, basic concepts, and definitions. Geochim. Cosmochim. Acta 1997, 61, 4935–4943. [Google Scholar] [CrossRef]
- Han, F.X.X.; Shiyab, S.; Chen, J.; Su, Y.; Monts, D.L.; Waggoner, C.A.; Matta, F.B. Extractability and bioavailability of mercury from a mercury sulfide contaminated soil in Oak Ridge, Tennessee, USA. Water Air Soil Pollut. 2008, 194, 67–75. [Google Scholar] [CrossRef]
- Mikac, N.; Foucher, D.; Niessen, S.; Fischer, J.C. Extractability of HgS (cinnabar and metacinnabar) by hydrochloric acid. Anal. Bioanal. Chem. 2002, 374, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.J.; Allen, H.E.; Li, Y.M.; Huang, C.P.; Sanders, P.F. Adsorption of mercury (II) by soil: Effects of pH, chloride, and organic matter. J. Environ. Qual. 1996, 25, 837–844. [Google Scholar] [CrossRef]
- Haitzer, M.; Aiken, G.R.; Ryan, J.N. Binding of mercury (II) to dissolved organic matter: The role of the mercury-to-DOM concentration ratio. Environ. Sci. Technol. 2002, 36, 3564–3570. [Google Scholar] [CrossRef] [PubMed]
- Borch, T.; Kretzschmar, R.; Kappler, A.; Van Cappellen, P.; Ginder-Vogel, M.; Voegelin, A.; Campbell, K. Biogeochemical redox processes and their impact on contaminant dynamics. Environ. Sci. Technol. 2010, 44, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Stumm, W.; Sulzberger, B. The cycling of iron in natural environments—Considerations based on laboratory studies of heterogeneous redox processes. Geochim. Cosmochim. Acta 1992, 56, 3233–3257. [Google Scholar] [CrossRef]
- Chorover, J.; Amistadi, M.K. Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces. Geochim. Cosmochim. Acta 2001, 65, 95–109. [Google Scholar] [CrossRef]
- Tipping, E.; Heaton, M.J. The adsorption of aquatic humic substances by two oxides of manganese. Geochim. Cosmochim. Acta 1983, 47, 1393–1397. [Google Scholar] [CrossRef]
- Avena, M.J.; Koopal, L.K. Desorption of humic acids from an iron oxide surface. Environ. Sci. Technol. 1998, 32, 2572–2577. [Google Scholar] [CrossRef]
- Grybos, M.; Davranche, M.; Gruau, G.; Petitjean, P.; Pedrot, M. Increasing pH drives organic matter solubilization from wetland soils under reducing conditions. Geoderma 2009, 154, 13–19. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilli, R.S.; Karlen, C.; Weber, M.; Rüegg, J.; Barmettler, K.; Biester, H.; Boivin, P.; Kretzschmar, R. Speciation and Mobility of Mercury in Soils Contaminated by Legacy Emissions from a Chemical Factory in the Rhône Valley in Canton of Valais, Switzerland. Soil Syst. 2018, 2, 44. https://doi.org/10.3390/soilsystems2030044
Gilli RS, Karlen C, Weber M, Rüegg J, Barmettler K, Biester H, Boivin P, Kretzschmar R. Speciation and Mobility of Mercury in Soils Contaminated by Legacy Emissions from a Chemical Factory in the Rhône Valley in Canton of Valais, Switzerland. Soil Systems. 2018; 2(3):44. https://doi.org/10.3390/soilsystems2030044
Chicago/Turabian StyleGilli, Robin Sue, Claudine Karlen, Mischa Weber, Johanna Rüegg, Kurt Barmettler, Harald Biester, Pascal Boivin, and Ruben Kretzschmar. 2018. "Speciation and Mobility of Mercury in Soils Contaminated by Legacy Emissions from a Chemical Factory in the Rhône Valley in Canton of Valais, Switzerland" Soil Systems 2, no. 3: 44. https://doi.org/10.3390/soilsystems2030044
APA StyleGilli, R. S., Karlen, C., Weber, M., Rüegg, J., Barmettler, K., Biester, H., Boivin, P., & Kretzschmar, R. (2018). Speciation and Mobility of Mercury in Soils Contaminated by Legacy Emissions from a Chemical Factory in the Rhône Valley in Canton of Valais, Switzerland. Soil Systems, 2(3), 44. https://doi.org/10.3390/soilsystems2030044