Sources and Pathways of Formation of Recalcitrant and Residual Phosphorus in an Agricultural Soil


Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Description and Soil Sampling
2.2. Soil Characterization
2.3. 31P Solid-State NMR Spectroscopy
2.4. Extraction of Soil Phosphorus Pools
2.5. Purification of Extracted Solution for Isotope Analyses
2.6. Measurement of Phosphate Oxygen Isotope Ratios
3. Results
3.1. Mineralogical Composition of Soils
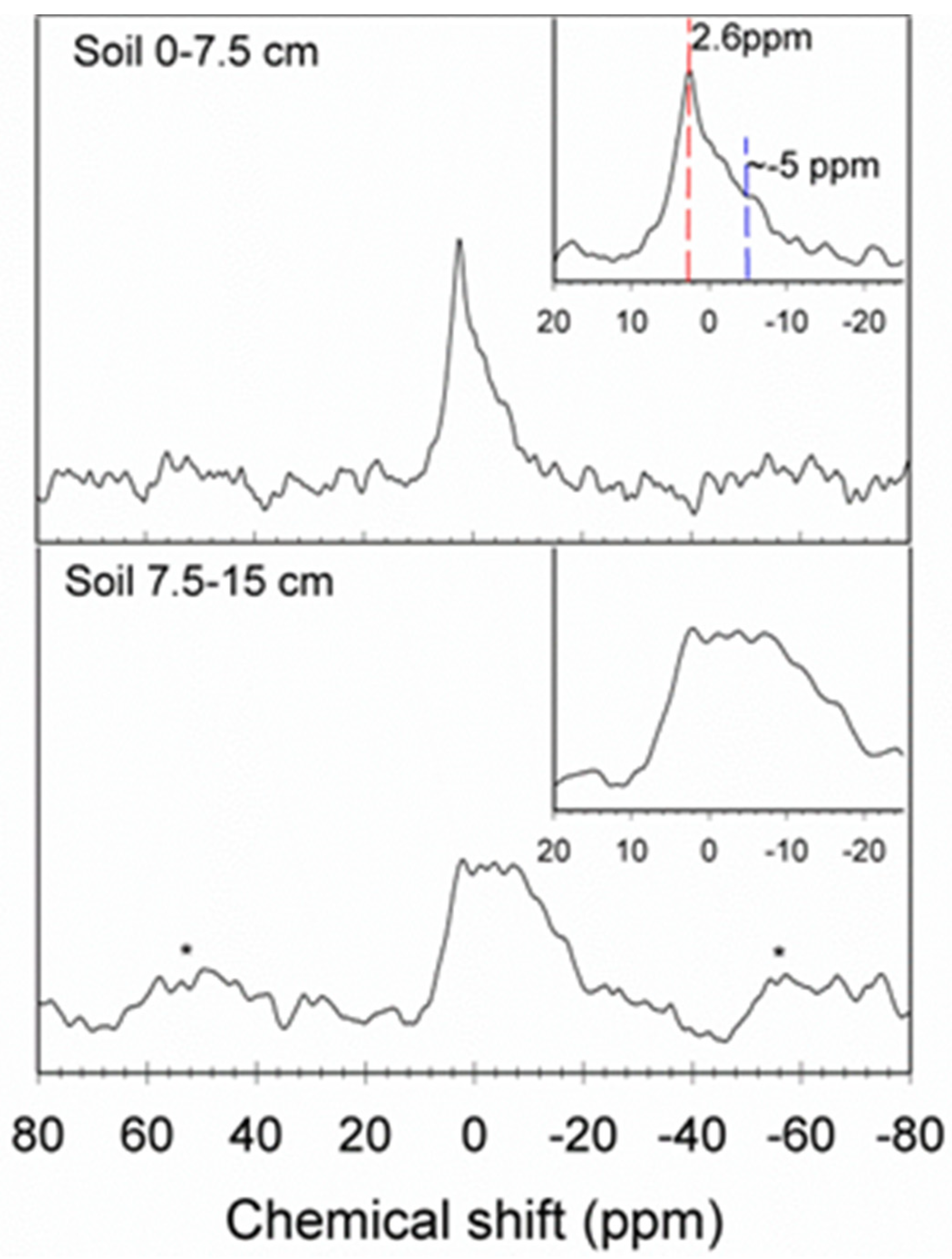
3.2. 31P NMR Results on the Composition of Phosphorus Minerals
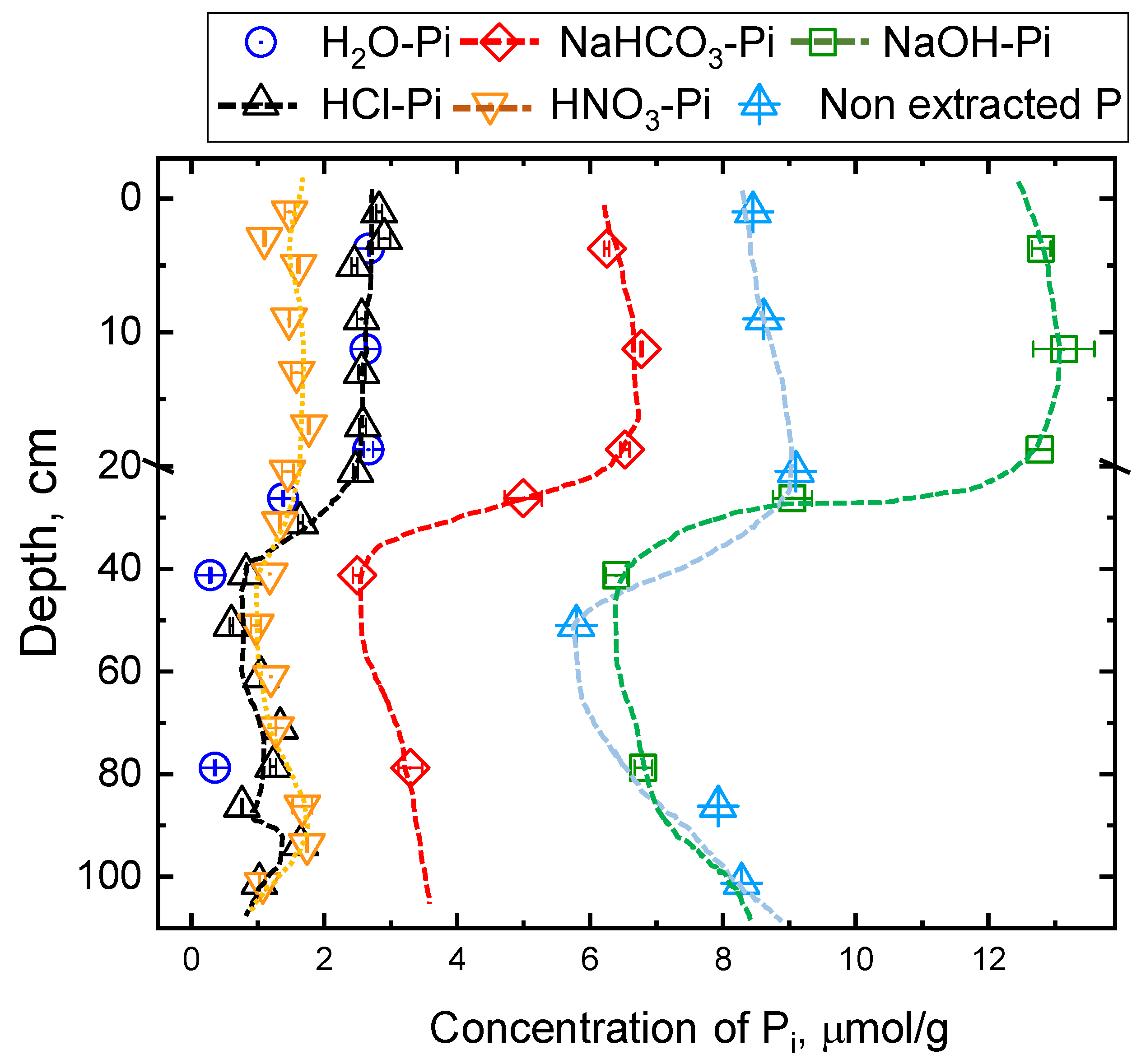
3.3. Concentrations and Isotopic Compositions of Phosphorus Pools
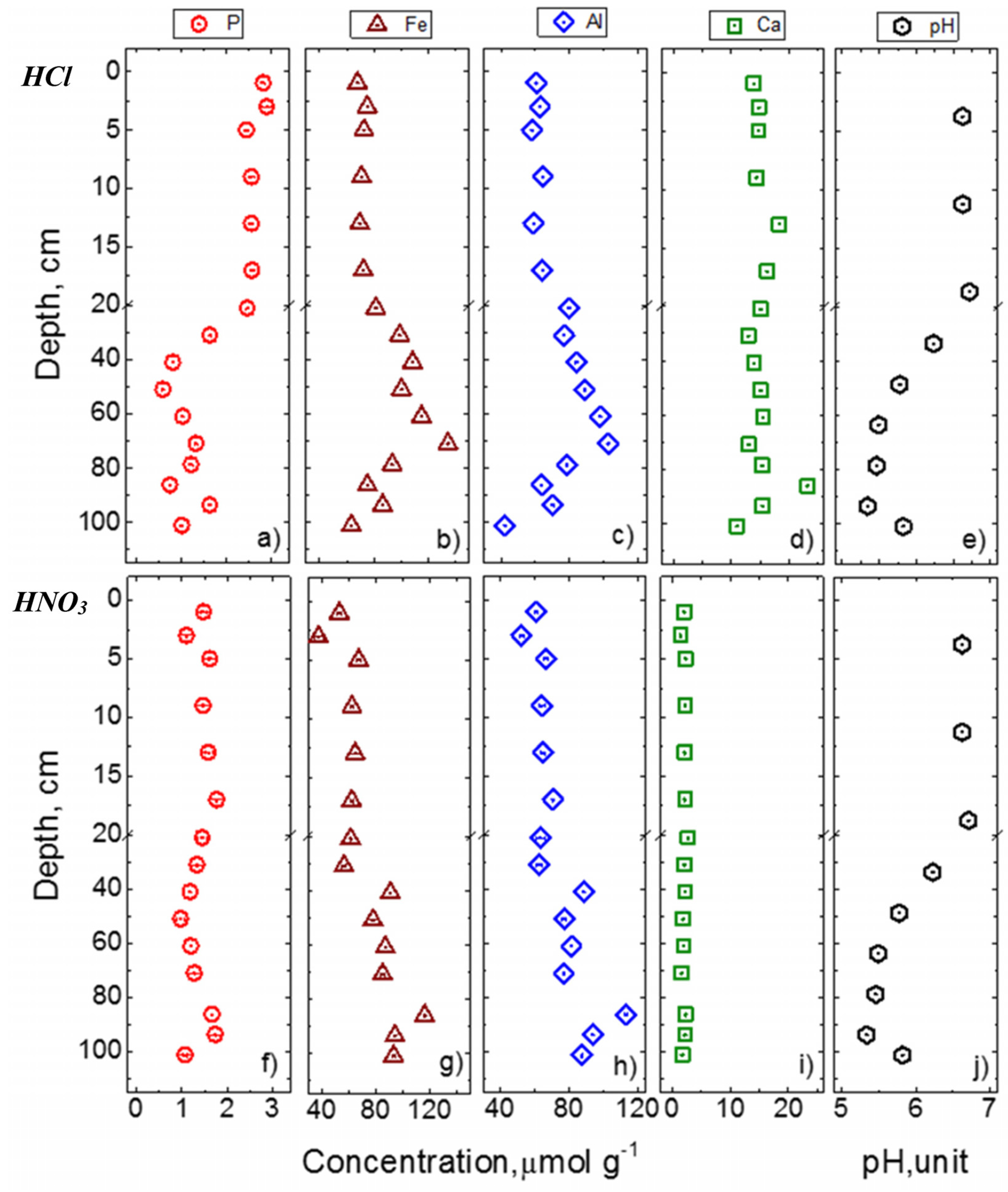
3.4. Concentrations of P, Fe, Al, and Ca in Acid Extracts
4. Discussion
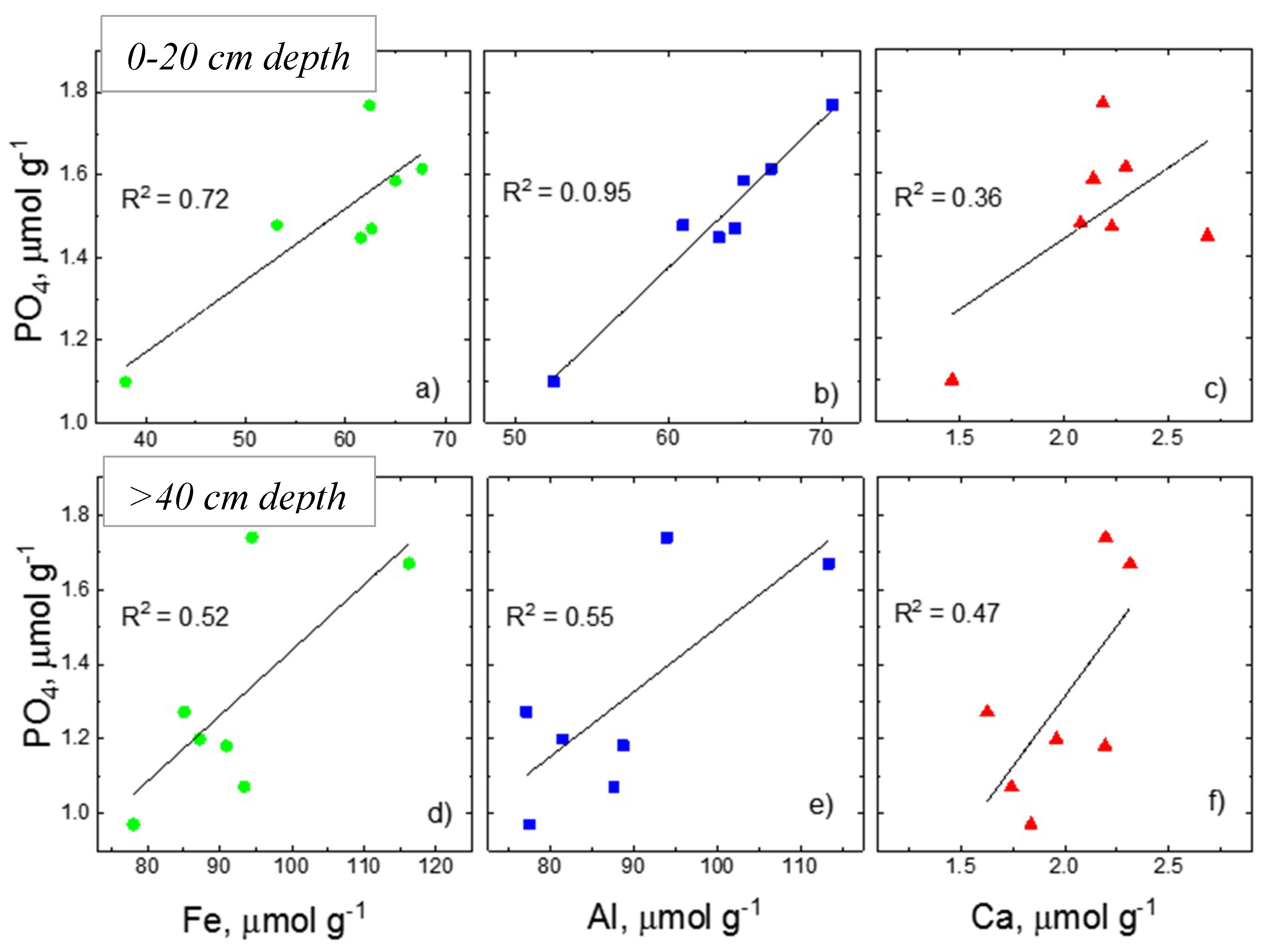
4.1. Potential Sources and Pathways Forming Acid-P Pools: Insights from Associated Elements
4.2. Potential Sources and Pathways Forming Acid-P Pools: Insights from Isotope Results
4.3. Vertical Movement of Phosphorus in Soil
5. Implications
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suh, S.; Yee, S. Phosphorus use-efficiency of agriculture and food system in the US. Chemosphere 2011, 84, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Cordell, D.; Drangert, J.-O.; White, S. The story of phosphorus: Global food security and food for thought. Glob. Environ. Chang. 2009, 19, 292–305. [Google Scholar] [CrossRef]
- Morse, D.; Head, H.H.; Wilcox, C.J. Disappearance of phosphorus in phytate from concentrates invitro and from rations fed to lactating dairy-cows. J. Dairy Sci. 1992, 75, 1979–1986. [Google Scholar] [CrossRef]
- Richardson, A.E.; Simpson, R.J. Soil microorganisms mediating phosphorus availability. Plant Physiol. 2011, 156, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Gaxiola, R.A.; Edwards, M.; Elser, J.J. A transgenic approach to enhance phosphorus use efficiency in crops as part of a comprehensive strategy for sustainable agriculture. Chemosphere 2011, 84, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Jaisi, D.P.; Blake, R.E. Advances in using oxygen isotope ratios of phosphate to understand phosphorus cycling in the environment. Adv. Agron. 2014, 125, 1–53. [Google Scholar]
- Kruse, J.; Abraham, M.; Amelung, W.; Baum, C.; Bol, R.; Kuhn, O.; Lewandowski, H.; Niederberger, J.; Oelmann, Y.; Ruger, C.; et al. Innovative methods in soil phosphorus research: A review. J. Plant Nutr. Soil Sci. 2015, 178, 43–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.C.; Jackson., M.L. Fractionation of soil phosphorus. Soil Sci. 1957, 84, 133–144. [Google Scholar] [CrossRef]
- Hedley, M.J.; Stewart, J.W.B.; Chauhan, B. Changes in inorganic and organic soil phosphorus fractions induced by cultivation practices and by laboratory incubations. Soil Sci. Soc. Am. J. 1982, 46, 970–976. [Google Scholar] [CrossRef]
- Tiessen, H.; Stewart, J.W.B.; Cole, C.V. Pathways of phosphorus transformations in soils of differing pedogenesis. Soil Sci. Soc. Am. J. 1984, 48, 853–858. [Google Scholar] [CrossRef]
- Barrow, N.J. Reaction of anions and cations with variable-charge soils. Adv. Agron. 1986, 38, 183–230. [Google Scholar]
- Ajiboye, B.; Akinremi, A.O.; Hu, Y.; Flaten, D.N. Phosphorus speciation of sequential extracts of organic amendments using nuclear magnetic resonance and x-ray absorption near-edge structure spectroscopies. J. Environ. Qual. 2007, 36, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.; Negassa, W.; Appathurai, N.; Zuin, L.; Leinweber, P. Phosphorus speciation in sequentially extracted agro-industrial by-products: Evidence from x-ray absorption near edge structure spectroscopy. J. Environ. Qual. 2010, 39, 2179–2184. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, Y.F.; Yang, J.J.; Abdi, D.; Cade-Menun, B.J. Investigation of soil legacy phosphorus transformation in long-term agricultural fields using sequential fractionation, p k-edge xanes and solution p nmr spectroscopy. Environ. Sci. Technol. 2015, 49, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Jaisi, D.P.; Blake, R.E. Tracing sources and cycling of phosphorus in peru margin sediments using oxygen isotopes in authigenic and detrital phosphates. Geochim. Cosmochim. Acta 2010, 74, 3199–3212. [Google Scholar] [CrossRef]
- Zohar, I.; Shaviv, A.; Young, M.; Kendall, C.; Silva, S.; Paytan, A. Phosphorus dynamics in soils irrigated with reclaimed waste water or fresh water—A study using oxygen isotopic composition of phosphate. Geoderma 2010, 159, 109–121. [Google Scholar] [CrossRef]
- Angert, A.; Weiner, T.; Mazeh, S.; Tamburini, F.; Frossard, E.; Bernasconi, S.M.; Sternberg, M. Seasonal variability of soil phosphate stable oxygen isotopes in rainfall manipulation experiments. Geochim. Cosmochim. Acta 2011, 75, 4216–4227. [Google Scholar] [CrossRef]
- Tamburini, F.; Pfahler, V.; Bunemann, E.K.; Guelland, K.; Bernasconi, S.M.; Frossard, E. Oxygen isotopes unravel the role of microorganisms in phosphate cycling in soils. Environ. Sci. Technol. 2012, 46, 5956–5962. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.R.; Li, X.N.; Jaisi, D.P. Transformation of phosphorus pools in an agricultural soil: An application of oxygen-18 labeling in phosphate. Soil Sci. Soc. Am. J. 2016, 80, 69–78. [Google Scholar] [CrossRef]
- Joshi, S.R.; Kukkadapu, R.K.; Burdige, D.J.; Bowden, M.E.; Sparks, D.L.; Jaisi, D.P. Organic matter remineralization predominates phosphorus cycling in the mid-bay sediments in the chesapeake bay. Environ. Sci. Technol. 2015, 49, 5887–5896. [Google Scholar] [CrossRef] [PubMed]
- Zohar, I.; Shaviv, A.; Klass, T.; Roberts, K.; Paytan, A. Method for the analysis of oxygen isotopic composition of soil phosphate fractions. Environ. Sci. Technol. 2010, 44, 7583–7588. [Google Scholar] [CrossRef] [PubMed]
- Johansen, H.S.; Middleboe, V.; Larsen, S. Delabelling of 18 o enriched phosphate added to soil as a function of biological activity in the soil. In Stable Isotopes in Plant Nutrition, Soil Fertility and Environmental Studies; IAEA: Vienna, Austria, 1991; pp. 553–559. [Google Scholar]
- Bauke, S.L.; von Sperber, C.; Tamburini, F.; Gocke, M.I.; Honermeier, B.; Schweitzer, K.; Baumecker, M.; Don, A.; Sandhage-Hofmann, A.; Amelung, W. Subsoil phosphorus is affected by fertilization regime in long-term agricultural experimental trials. Eur. J. Soil Sci. 2018, 69, 103–112. [Google Scholar] [CrossRef]
- Bauke, S.L.; von Sperber, C.; Siebers, N.; Tamburini, F.; Amelung, W. Biopore effects on phosphorus biogeochemistry in subsoils. Soil Biol. Biochem. 2017, 111, 157–165. [Google Scholar] [CrossRef]
- Von Sperber, C.; Stallforth, R.; Du Preez, C.; Amelung, W. Changes in soil phosphorus pools during prolonged arable cropping in semiarid grasslands. Eur. J. Soil Sci. 2017, 68, 462–471. [Google Scholar] [CrossRef]
- O’Neil, J.R.; Vennemann, T.W.; McKenzie, W.F. Effects of speciation on equilibrium fractionations and rates of oxygen isotope exchange between (PO4) aq and H2O. Geochim. Cosmochim. Acta 2003, 67, 3135–3144. [Google Scholar] [CrossRef]
- Lecuyer, C.; Grandjean, P.; Sheppard, S.M.F. Oxygen isotope exchange between dissolved phosphate and water at temperatures ≤135 c: Inorganic versus biological fractionations. Geochim. Cosmochim. Acta 1999, 63, 855–862. [Google Scholar] [CrossRef]
- Jaisi, D.P.; Blake, R.E.; Kukkadapu, R.K. Fractionation of oxygen isotopes in phosphate during its interactions with iron oxides. Geochim. Cosmochim. Acta 2010, 74, 1309–1319. [Google Scholar] [CrossRef]
- Stout, L.M.; Joshi, S.R.; Kana, T.M.; Jaisi, D.P. Microbial activities and phosphorus cycling: An application of oxygen isotope ratios in phosphate. Geochim. Cosmochim. Acta 2014, 138, 101–116. [Google Scholar] [CrossRef]
- Longinelli, A.; Nuti, S. Revised phosphate-water isotopic temperature scale. Earth Planet. Sci. Lett. 1973, 19, 373–376. [Google Scholar] [CrossRef]
- Blake, R.E.; O’Neil, J.R.; Garcia, G.A. Oxygen isotope systematics of biologically mediated reactions of phosphate: I. Microbial degradation of organophosphorus compounds. Geochim. Cosmochim. Acta 1997, 61, 4411–4422. [Google Scholar] [CrossRef]
- Ruttenberg, K.C. Development of a sequential extraction method for different forms of phosphorus in marine sediments. Limnol. Oceanogr. 1992, 37, 1460–1482. [Google Scholar] [CrossRef] [Green Version]
- Bish, D.L.; Howard, S.A. Quantitative phase-analysis using the rietveld method. J. Appl. Crystallogr. 1988, 21, 86–91. [Google Scholar] [CrossRef]
- Murphy, J.; Riley, J.P. A modified single solution method for the determination of phosphate in natural water. Anal. Chem. Acta 1962, 27, 31–36. [Google Scholar] [CrossRef]
- Rowland, A.P.; Haygarth, P.M. Determination of total dissolved phosphorus in soil solutions. J. Environ. Qual. 1997, 26, 410–415. [Google Scholar] [CrossRef]
- Jaisi, D.P.; Blake, R.E.; Liang, Y.; Chan, S.-J. Investigation of compound-specific organic-inorganic phos-phorus transformation using stable isotope ratios in phosphate. In Applied Manure and Nutrient Chemistry for Sustainable Agriculture of Environment; He, Z., Zhang, H., Eds.; Springer Science + Business Media: Dordrecht, The Netherlands, 2014; pp. 267–292. [Google Scholar]
- Karl, D.M.; Tien, G. Magic: A sensitive and precise method for measuring dissolved phosphorus in aquatic environments. Limnol. Oceanogr. 1992, 37, 105–116. [Google Scholar] [CrossRef]
- Chang, S.J.; Blake, R.E. Precise calibration of equilibrium oxygen isotope fractionations between dissolved phosphate and water from 3 to 37 degrees c. Geochim. Cosmochim. Acta 2015, 150, 314–329. [Google Scholar] [CrossRef]
- Lehmann, J.; Lan, Z.; Hyland, C.; Sato, S.; Solomon, D.; Ketterings, Q.M. Long-term dynamics of phosphorus forms and retention in manure-amended soils. Environ. Sci. Technol. 2005, 39, 6672–6680. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, R.H.; Stewart, J.W.B.; Dormaar, J.F.; Schaalje, G.B. Long-term crop-rotation and fertilizer effects on phosphorus transformations. 2. In a luvisolic soil. Can. J. Soil Sci. 1992, 72, 581–589. [Google Scholar] [CrossRef]
- Richards, J.E.; Bates, T.E.; Sheppard, S.C. Changes in the forms and distribution of soil-phosphorus due to long-term corn production. Can. J. Soil Sci. 1995, 75, 311–318. [Google Scholar] [CrossRef]
- Guo, F.; Yost, R.S.; Hue, N.V.; Evensen, C.I.; Silva, J.A. Changes in phosphorus fractions in soils under intensive plant growth. Soil Sci. Soc. Am. J. 2000, 64, 1681–1689. [Google Scholar] [CrossRef]
- Cross, A.F.; Schlesinger, W.H. A literature review and evaluation of the. Hedley fractionation: Applications to the biogeochemical cycle of soil phosphorus in natural ecosystems. Geoderma 1995, 64, 197–214. [Google Scholar] [CrossRef]
- Tiessen, H.; Stewart, J.W.B.; Moir, J.O. Changes in organic and inorganic phosphorus composition of 2 grassland soils and their particle-size fractions during 60–90 years of cultivation. J. Soil Sci. 1983, 34, 815–823. [Google Scholar] [CrossRef]
- McLaren, T.I.; Guppy, C.N.; Tighe, M.K.; Schefe, C.R.; Flavel, R.J.; Cowie, B.C.C.; Tadich, A. Validation of soil phosphate removal by alkaline and acidic reagents in a vertosol soil using xanes spectroscopy. Commun. Soil Sci. Plant Anal. 2015, 46, 1998–2017. [Google Scholar] [CrossRef]
- Zhang, T.Q.; MacKenzie, A.F. Changes of phosphorous fractions under continuous corn production in a temperate clay soil. Plant Soil 1997, 192, 133–139. [Google Scholar] [CrossRef]
- Angert, A.; Weiner, T.; Mazeh, S.; Sternberg, M. Soil phosphate stable oxygen isotopes across rainfall and bedrock gradients. Environ. Sci. Technol. 2012, 46, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Ayliffe, L.K.; Lister, A.M.; Chivas, A.R. The preservation of glacial-interglacial climatic signatures in the oxygen isotopes of elephant skeletal phosphate. Palaeogeogr. Palaeoclimatol. Palaeoecol. 1992, 99, 179–191. [Google Scholar] [CrossRef]
- Mizota, C.; Domon, Y.; Yoshida, N. Oxygen isotope composition of natural phosphates from volcanic ash soils of the great rift valley of Africa and east Java, Indonesia. Geoderma 1992, 53, 111–123. [Google Scholar] [CrossRef]
- Syers, J.K.; Johnson, A.E.; Curtin, D. Efficiency of soil and fertilizer phosphorus use: Reconciling changing concepts of soil phosphorus behavior with agronomic information. In FAO Fertilizer and Plant Nutrition Bulletin; Food and Agriculture Organization of the United Nations: Rome, Italy, 2008. [Google Scholar]
- Arai, Y.; Sparks, D.L. Phosphate reaction dynamics in soils and soil components: A multiscale approach. Adv. Agron. 2007, 94, 135–179. [Google Scholar]
- Bolan, N.S.; Barrow, N.J.; Posner., A.M. Describing the effect of time on sorption of phosphate by iron and aluminum hydroxy-oxides. J. Soil Sci. 1985, 36, 187–197. [Google Scholar] [CrossRef]
- Parfitt, R.L. Phosphate reactions with natural allophane, ferrihydrite, and goethite. J. Soil Sci. 1989, 40, 359–369. [Google Scholar] [CrossRef]
- Sims, J.T.; Simard, R.R.; Joern, B.C. Phosphorus loss in agricultural drainage: Historical perspective and current research. J. Environ. Qual. 1998, 27, 277–293. [Google Scholar] [CrossRef]
- Kleinman, P.J.A.; Church, C.; Saporito, L.S.; McGrath, J.M.; Reiter, M.S.; Allen, A.L.; Tingle, S.; Binford, G.D.; Han, K.; Joern, B.C. Phosphorus leaching from agricultural soils of the Delmarva Peninsula, USA. J. Environ. Qual. 2015, 44, 524–534. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, L.W.; Moldrup, P.; Rubæk, G.H.; Schelde, K.; Djurhuus, J. Particle leaching and particle-facilitated transport of phosphorus at field scale. Vadose Zone J. 2004, 3, 462–470. [Google Scholar] [CrossRef]
- Djodjic, F.; Ulen, B.; Bergstrom, L. Temporal and spatial variations of phosphorus losses and drainage in a structured clay soil. Water Res. 2000, 34, 1687–1695. [Google Scholar] [CrossRef]
- Jiang, X.; Bol, R.; Nischwitz, V.; Siebers, N.; Willbold, S.; Vereecken, H.; Amelung, W.; Klumpp, E. Phosphorus containing water dispersible nanoparticles in arable soil. J. Environ. Qual. 2015, 44, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, G.; Ngo, P.T.; Rumpel, C.; Calabi-Floody, M.; Redel, Y.; Turner, B.L.; Condron, L.M.; Mora, M.D. Chemical nature of residual phosphorus in andisols. Geoderma 2015, 271, 27–31. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Depth | Quartz | Mica | Feldspar | Kaolinite | Amorphous | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NaOH | HCl | HNO3 | NaOH | HCl | HNO3 | NaOH | HCl | HNO3 | NaOH | HCl | HNO3 | NaOH | HCl | HNO3 | |
| (cm) | % | % | % | % | % | ||||||||||
| 0−2 | 66 | 68 | 54 | 9 | 10 | 7 | 18 | 14 | 12 | 7 | 5 | 3 | ¥ | 3 | 25 |
| 8−10 | 69 | 64 | 56 | 12 | 9 | 8 | 14 | 19 | 17 | 5 | 7 | 3 | ¥ | 2 | 16 |
| 20−22 | 67 | 61 | 59 | 8 | 10 | 7 | 15 | 15 | 13 | 4 | 6 | 4 | 5 | 8 | 16 |
| 50−52 | 61 | 62 | 46 | 11 | 11 | 9 | 18 | 14 | 13 | 7 | 7 | 4 | 3 | 7 | 28 |
| 82.5−90 | 63 | 58 | 44 | 12 | 11 | 9 | 14 | 13 | 13 | 7 | 8 | 4 | 4 | 10 | 30 |
| 97.5−105 | 57 | 61 | 53 | 8 | 10 | 6 | 10 | 10 | 8 | 5 | 6 | 4 | 3 | 13 | 29 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, S.R.; Li, W.; Bowden, M.; Jaisi, D.P. Sources and Pathways of Formation of Recalcitrant and Residual Phosphorus in an Agricultural Soil. Soil Syst. 2018, 2, 45. https://doi.org/10.3390/soilsystems2030045
Joshi SR, Li W, Bowden M, Jaisi DP. Sources and Pathways of Formation of Recalcitrant and Residual Phosphorus in an Agricultural Soil. Soil Systems. 2018; 2(3):45. https://doi.org/10.3390/soilsystems2030045
Chicago/Turabian StyleJoshi, Sunendra R., Wei Li, Mark Bowden, and Deb P. Jaisi. 2018. "Sources and Pathways of Formation of Recalcitrant and Residual Phosphorus in an Agricultural Soil" Soil Systems 2, no. 3: 45. https://doi.org/10.3390/soilsystems2030045
APA StyleJoshi, S. R., Li, W., Bowden, M., & Jaisi, D. P. (2018). Sources and Pathways of Formation of Recalcitrant and Residual Phosphorus in an Agricultural Soil. Soil Systems, 2(3), 45. https://doi.org/10.3390/soilsystems2030045