Products of Hexavalent Chromium Reduction by Green Rust Sodium Sulfate and Associated Reaction Mechanisms

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
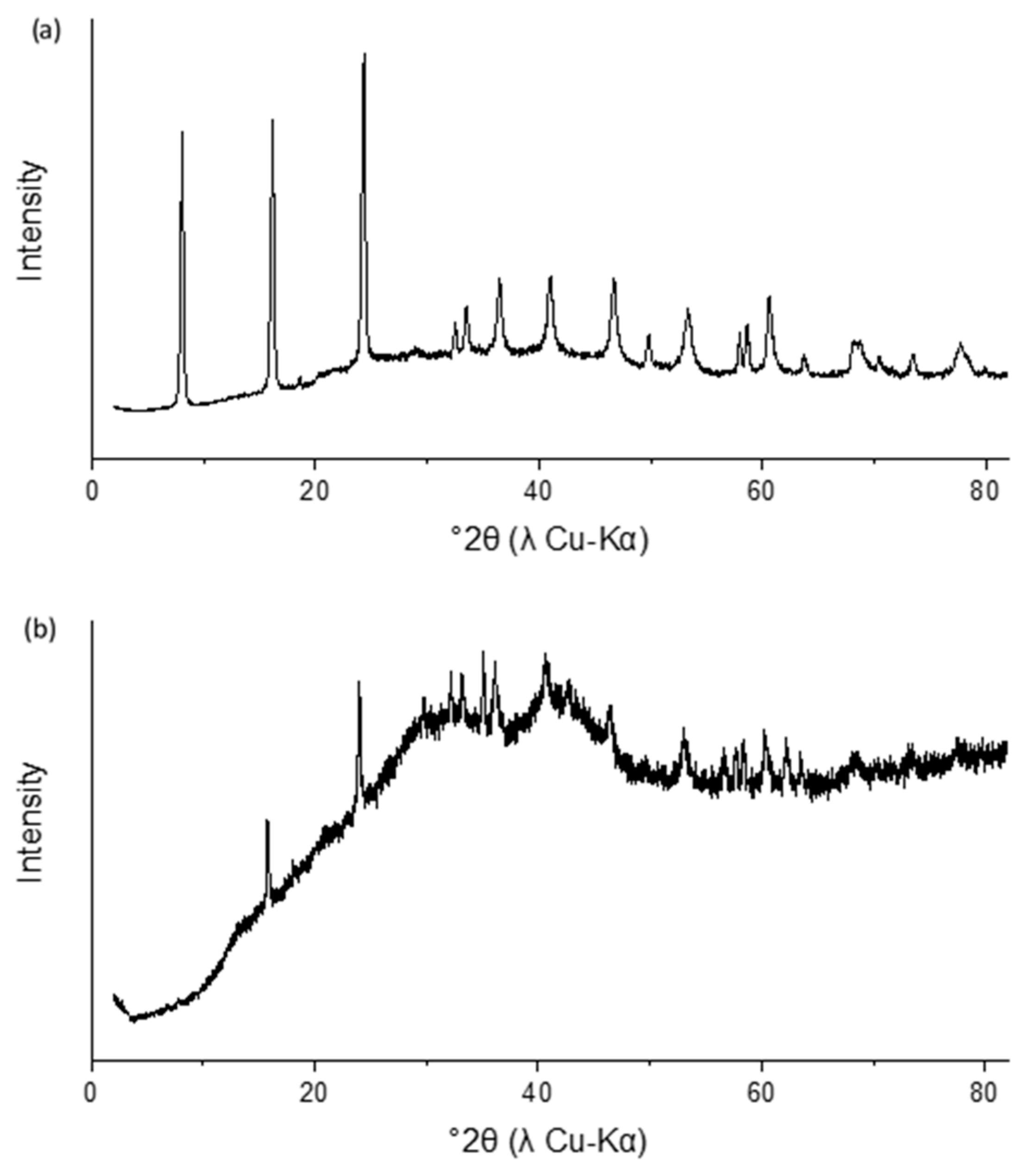
3.1. Characterization and Stability of Sulfate Green Rust
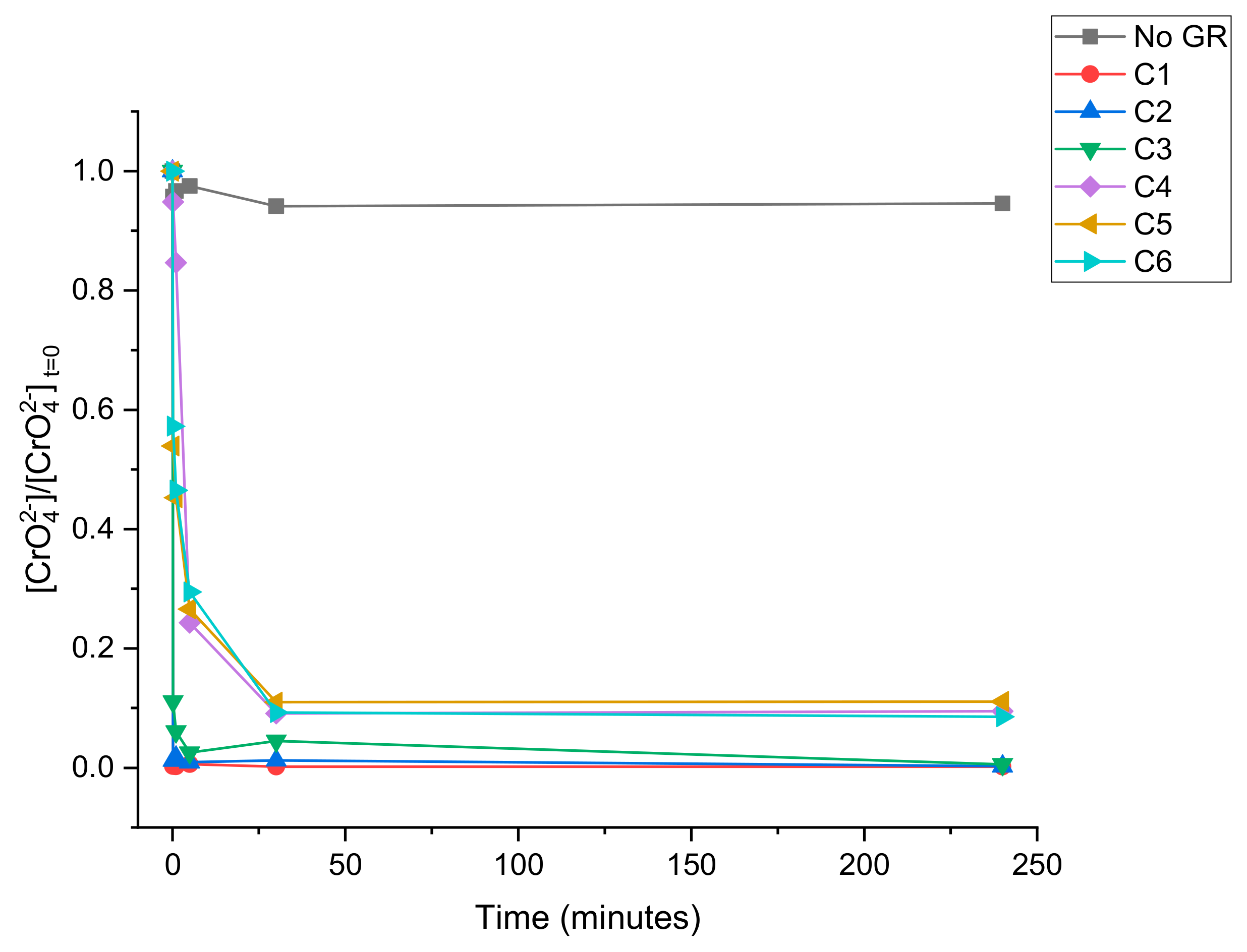
3.2. CrO42− Reduction Kinetics
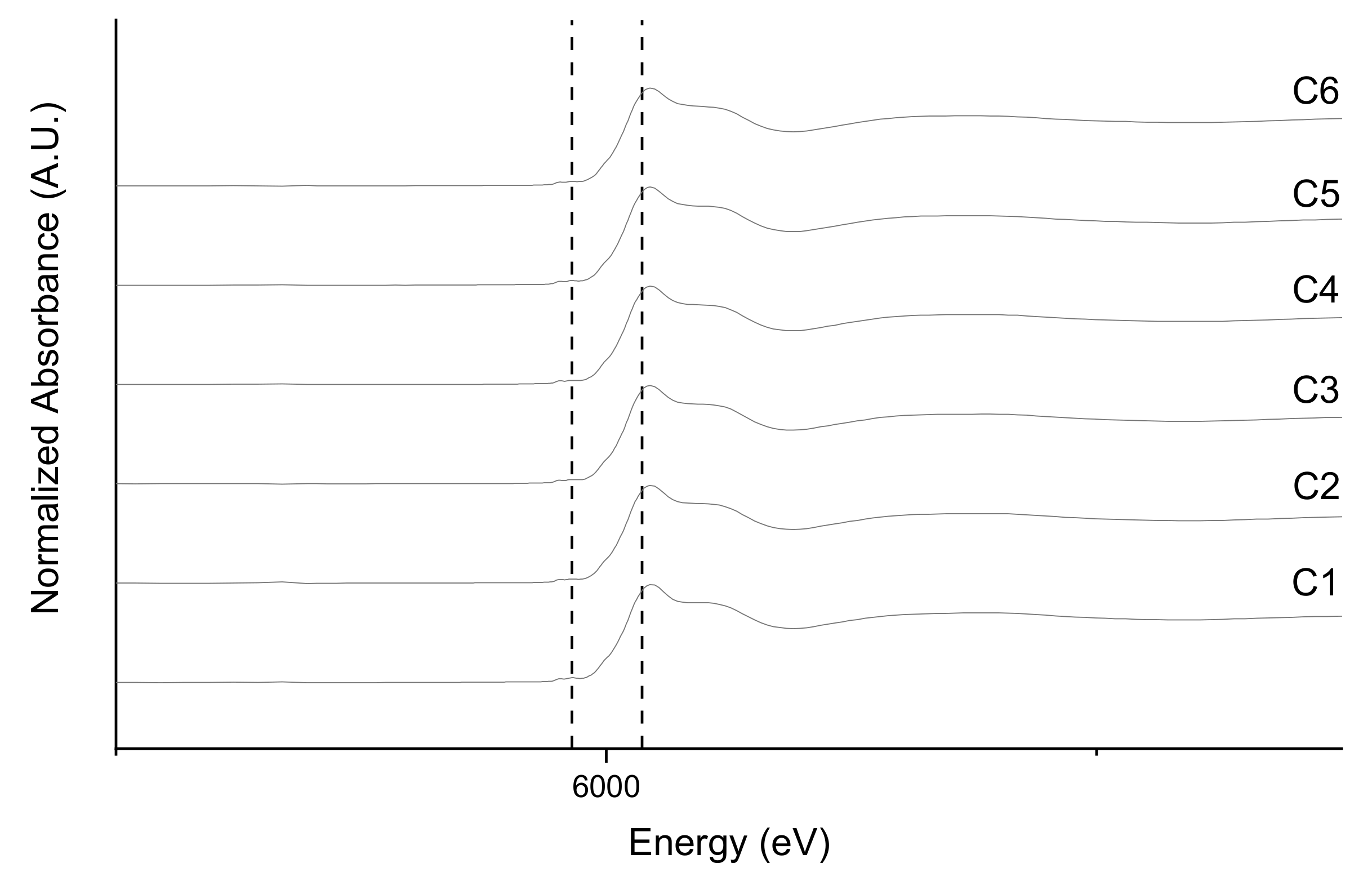
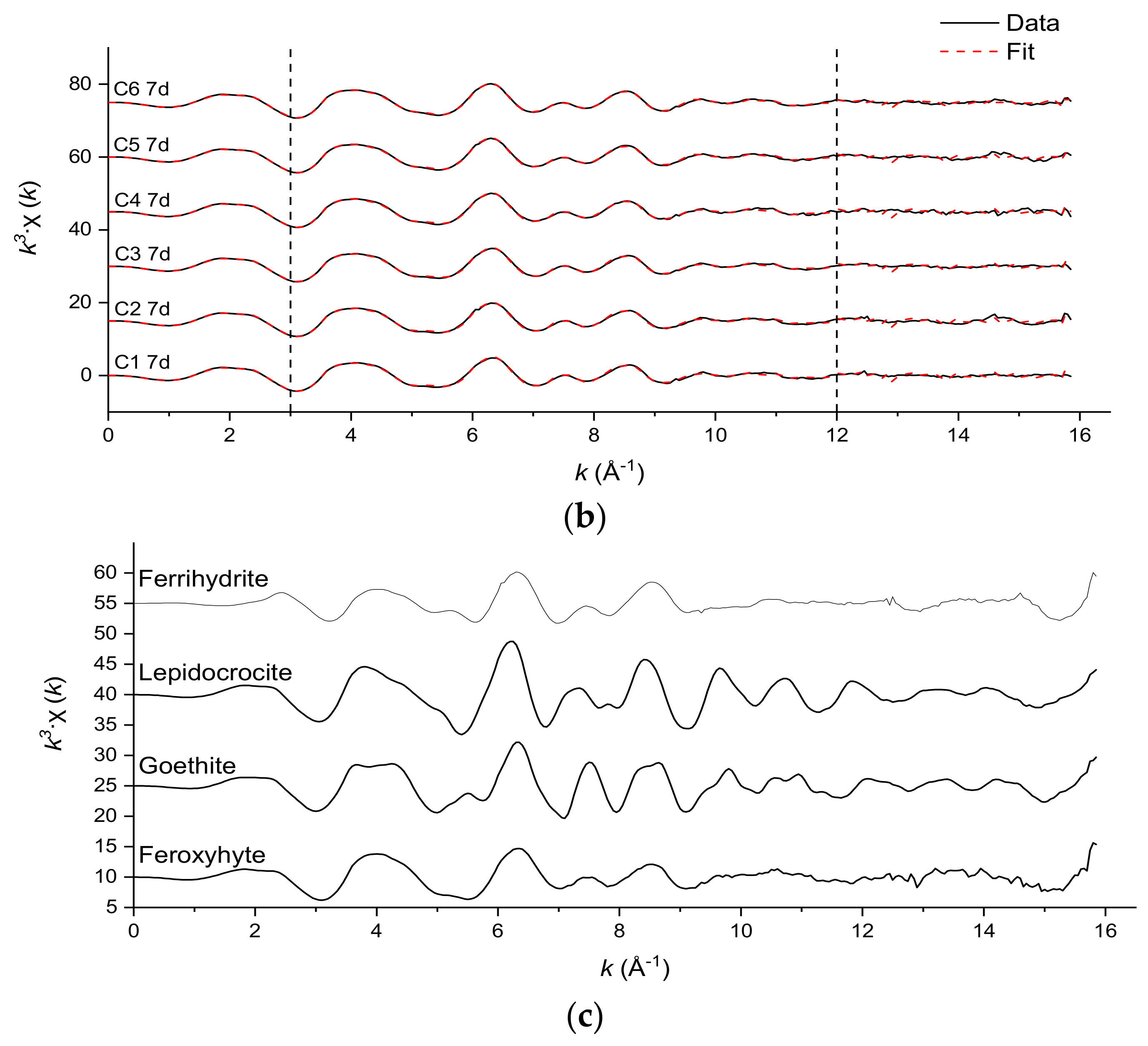
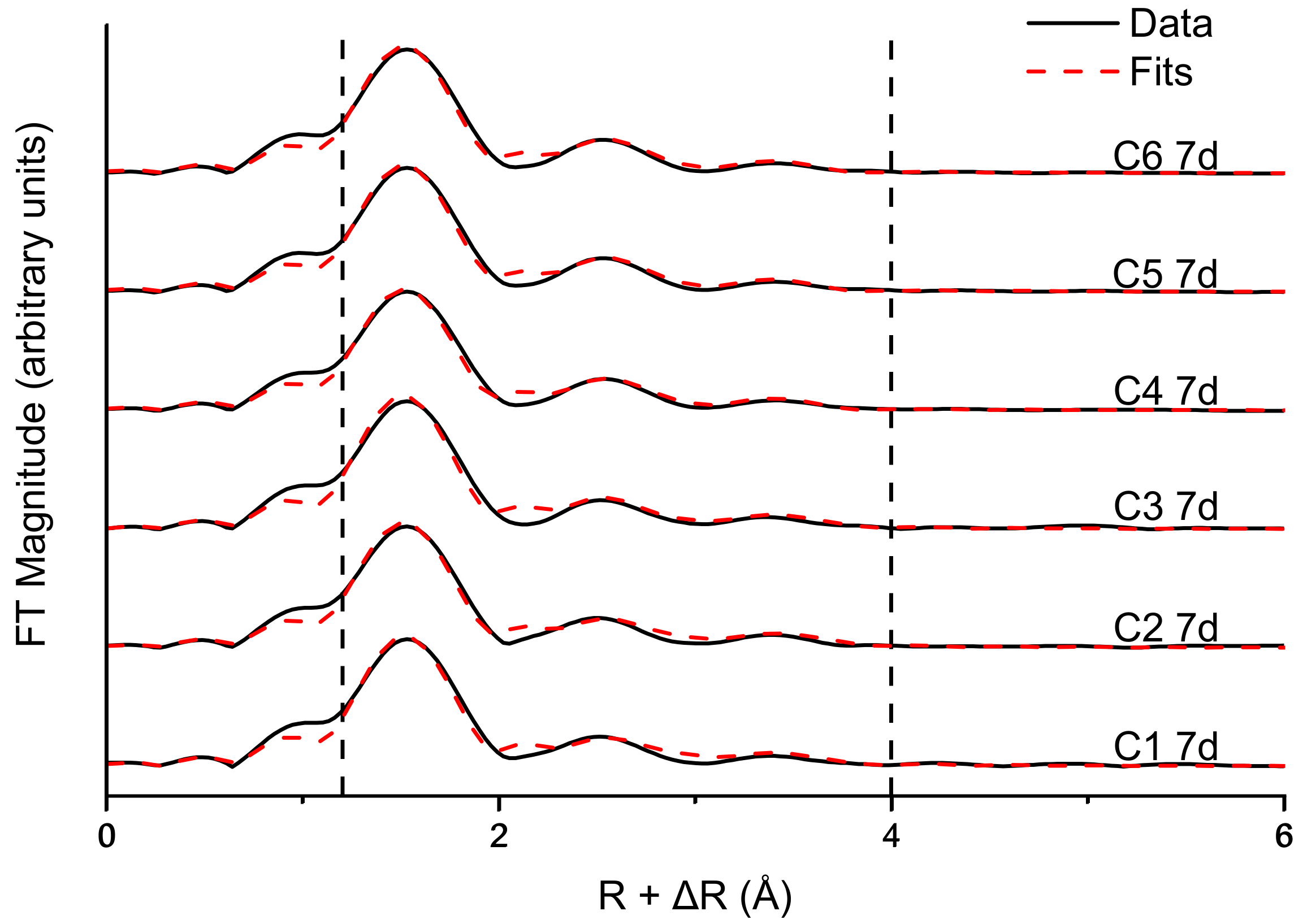
3.3. Fe and Cr Speciation
3.4. Electron Microscopy
4. Discussion
4.1. Kinetics of CrO42− Reduction by Green Rust Sulfate
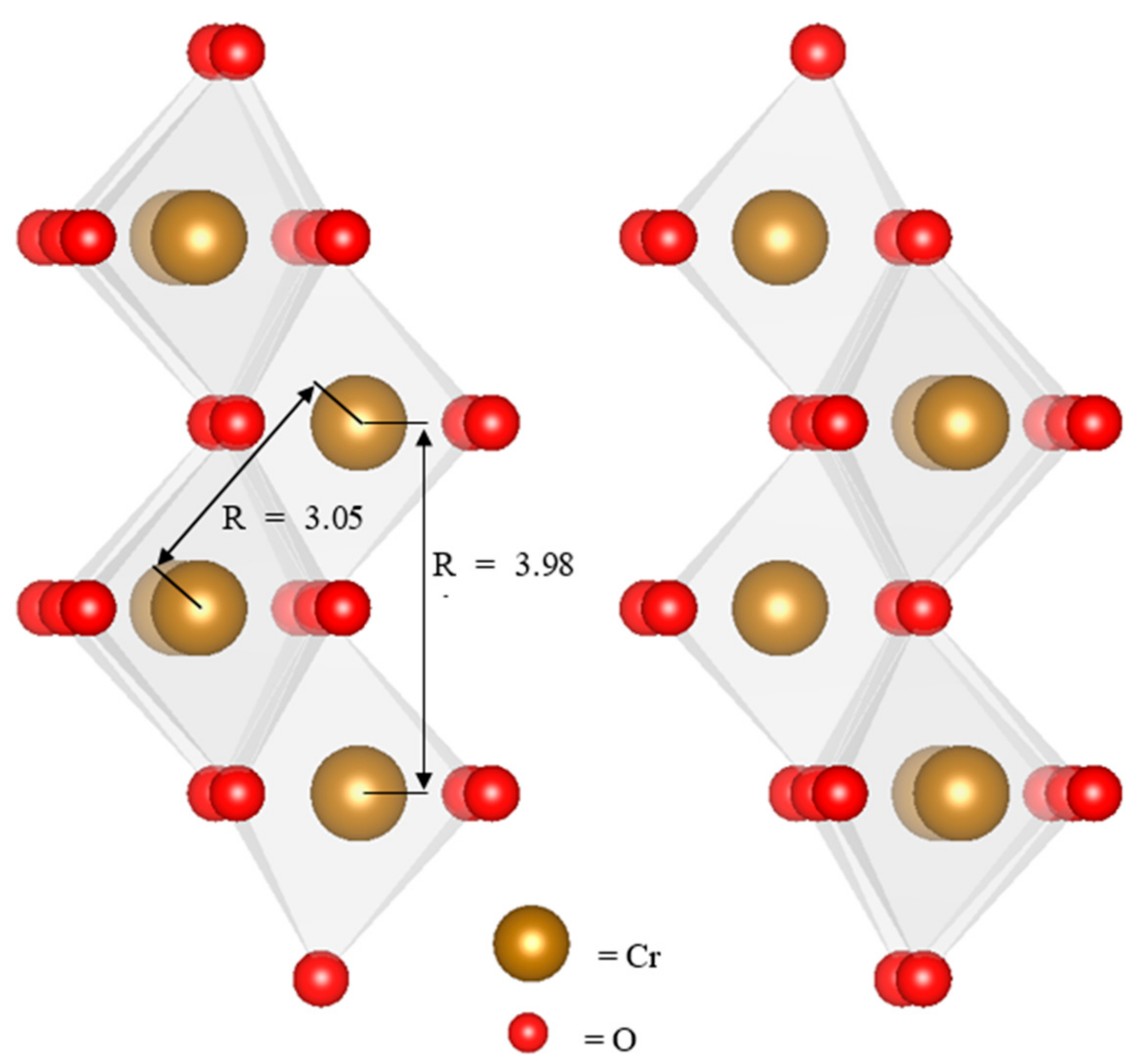
4.2. Composition of Observed Reaction Products
4.3. Reaction Mechanisms
4.4. Implications for Use in Permeable Reactive Barriers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saha, R.; Nandi, R.; Saha, B. Sources and toxicity of hexavalent chromium. J. Coord. Chem. 2011, 64, 1782–1806. [Google Scholar] [CrossRef]
- Barceloux, D.G. Chromium. J. Toxicol. Clin. Toxicol. 1999, 37, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Rai, D.; Sass, B.M.; Moore, D.A. Chromium(III) Hydrolysis Constants and Solubility of Chromium(III) Hydroxide. Inorg. Chem. 1987, 26, 345–349. [Google Scholar] [CrossRef]
- Rai, D.; Moore, D.A.; Hess, N.J.; Rosso, K.M.; Rao, L.; Heald, S.M. Chromium(III) Hydroxide Solubility in the Aqueous K+-H+-OH−-CO2-HCO3−-CO32−-H2O System: A Thermodynamic Model. J. Solut. Chem. 2007, 36, 1261–1285. [Google Scholar] [CrossRef]
- Su, C.; Ludwig, R.D. Treatment of Hexavalent Chromium in Chromite Ore Processing Solid Waste Using a Mixed Reductant Solution of Ferrous Sulfate and Sodium Dithionite. Environ. Sci. Technol. 2005, 39, 6208–6216. [Google Scholar] [CrossRef] [PubMed]
- Puls, R.W.; Paul, C.J.; Powell, R.M. The application of in situ permeable reactive (zero-valent iron) barrier technology for the remediation of chromate-contaminated groundwater: A field test. Appl. Geochem. 1999, 14, 989–1000. [Google Scholar] [CrossRef]
- Wilkin, R.T.; Su, C.; Ford, R.G.; Paul, C.J. Chromium-Removal Processes during Groundwater Remediation by a Zerovalent Iron Permeable Reactive Barrier. Environ. Sci. Technol. 2005, 39, 4599–4605. [Google Scholar] [CrossRef] [PubMed]
- Kumpiene, J.; Ore, S.; Renella, G.; Mench, M.; Lagerkvist, A.; Maurice, C. Assessment of zerovalent iron for stabilization of chromium, copper, and arsenic in soil. Environ. Pollut. 2006, 144, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Hausladen, D.M.; Fendorf, S. Hexavalent Chromium Generation within Naturally Structured Soils and Sediments. Environ. Sci. Technol. 2017, 51, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Liu, H.; Catalano, J.G.; Qian, A.; Wang, Z.; Giammar, D.E. Rates of Cr(VI) Generation from CrxFe1−x(OH)3 Solids upon Reaction with Manganese Oxide. Environ. Sci. Technol. 2017, 51, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Varadharajan, C.; Beller, H.R.; Bill, M.; Brodie, E.L.; Conrad, M.E.; Han, R.; Irwin, C.; Larsen, J.T.; Lim, H.C.; Molins, S.; et al. Reoxidation of Chromium(III) Products Formed under Different Biogeochemical Regimes. Environ. Sci. Technol. 2017, 51, 4918–4927. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, B.C.; Dideriksen, K.; Katz, A.; Nedel, S.; Bovet, N.; Sorensen, H.O.; Frandsen, C.; Gundlach, C.; Andersson, M.P.; Stipp, S.L. Incorporation of monovalent cations in sulfate green rust. Inorg. Chem. 2014, 53, 8887–8894. [Google Scholar] [CrossRef] [PubMed]
- Trolard, F.; Bourrié, G.; Abdelmoula, M.; Refait, P.; Feder, F. Fougerite, a new mineral of the pyroaurite-iowaite group: Description and crystal structure. Clays Clay Miner. 2007, 55, 323–334. [Google Scholar] [CrossRef]
- Lewis, D.G. Factors influencing the stability and properties of green rusts. Adv. Geoecol. 1997, 30, 345–372. [Google Scholar]
- Roh, Y.; Lee, S.Y.; Elless, M.P. Characterization of corrosion products in the permeable reactive barriers. Environ. Geol. 2000, 40, 184–194. [Google Scholar] [CrossRef]
- Refait, P.; Memet, J.B.; Bon, C.; Sabot, R.; Génin, J.M.R. Formation of the Fe(II)–Fe(III) hydroxysulphate green rust during marine corrosion of steel. Corros. Sci. 2003, 45, 833–845. [Google Scholar] [CrossRef]
- Su, C.; Puls, R.W. Significance of Iron(II,III) Hydroxycarbonate Green Rust in Arsenic Remediation Using Zerovalent Iron in Laboratory Column Tests. Environ. Sci. Technol. 2004, 38, 5224–5231. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, J.; Sherman, D.M. Sorption of As(III) and As(V) to siderite, green rust (fougerite) and magnetite: Implications for arsenic release in anoxic groundwaters. Chem. Geol. 2008, 255, 173–181. [Google Scholar] [CrossRef]
- Perez, J.P.H.; Freeman, H.M.; Schuessler, J.A.; Benning, L.G. The interfacial reactivity of arsenic species with green rust sulfate (GRSO4). Sci. Total Environ. 2019, 648, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H. Kinetics of nitrate reduction by green rusts—Effects of interlayer anion and Fe(II):Fe(III) ratio. Appl. Clay Sci. 2001, 18, 81–91. [Google Scholar] [CrossRef]
- Hansen, H.C.B.; Borggaard, O.K.; Sørensen, J. Evaluation of the free energy of formation of Fe(II)-Fe(III) hydroxide-sulphate (green rust) and its reduction of nitrite. Geochim. Cosmochim. Acta 1994, 58, 2599–2608. [Google Scholar] [CrossRef]
- Myneni, S.C. Abiotic Selenium Redox Transformations in the Presence of Fe(II,III) Oxides. Science 1997, 278, 1106–1109. [Google Scholar] [CrossRef]
- Borsig, N.; Scheinost, A.C.; Shaw, S.; Schild, D.; Neumann, T. Retention and multiphase transformation of selenium oxyanions during the formation of magnetite via iron(II) hydroxide and green rust. Dalton Trans. 2018, 47, 11002–11015. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.J.; Kelly, S.D.; Cook, R.E.; Csencsits, R.; Kemner, K.M. Reduction of Uranium(VI) by Mixed Iron(II)/Iron(III) Hydroxide (Green Rust): Formation of UO2Nanoparticles. Environ. Sci. Technol. 2003, 37, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, B.C.; Geckeis, H.; Marquardt, C.M.; Bauer, A.; Römer, J.; Wiss, T.; Schild, D.; Stipp, S.L.S. Neptunyl (Np) interaction with green rust. Geochim. Cosmochim. Acta 2011, 75, 1216–1226. [Google Scholar] [CrossRef]
- Loyaux-Lawniczak, S.; Refait, P.; Lecomte, P.; Ehrhardt, J.J.; Génin, J.M.R. The reduction of chromate ions by Fe(II) layered hydroxides. Hydrol. Earth Syst. Sci. 1999, 3, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Loyaux-Lawniczak, S.; Refait, P.; Ehrhardt, J.-J.; Lecomte, P.; Génin, J.-M.R. Trapping of Cr by Formation of Ferrihydrite during the Reduction of Chromate Ions by Fe(II)−Fe(III) Hydroxysalt Green Rusts. Environ. Sci. Technol. 2000, 34, 438–443. [Google Scholar] [CrossRef]
- Bond, D.L.; Fendorf, S. Kinetics and Structural Constraints of Chromate Reduction by Green Rusts. Environ. Sci. Technol. 2003, 37, 2750–2757. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.G.B.; Scherer, M.M. Kinetics of Cr(VI) Reduction by Carbonate Green Rust. Environ. Sci. Technol. 2001, 35, 3488–3494. [Google Scholar] [CrossRef] [PubMed]
- Legrand, L.; El Figuigui, A.; Mercier, F.; Chausse, A. Reduction of Aqueous Chromate by Fe(II)/Fe(III) Carbonate Green Rust: Kinetic and Mechanistic Studies. Environ. Sci. Technol. 2004, 38, 4587–4595. [Google Scholar] [CrossRef] [PubMed]
- Skovbjerg, L.L.; Stipp, S.L.S.; Utsunomiya, S.; Ewing, R.C. The mechanisms of reduction of hexavalent chromium by green rust sodium sulphate: Formation of Cr-goethite. Geochim. Cosmochim. Acta 2006, 70, 3582–3592. [Google Scholar] [CrossRef]
- Géhin, A.; Ruby, C.; Abdelmoula, M.; Benali, O.; Ghanbaja, J.; Refait, P.; Génin, J.-M.R. Synthesis of Fe(II-III) hydroxysulphate green rust by coprecipitation. Solid State Sci. 2002, 4, 61–66. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA and ARTEMIS Interactive Graphical Data Analysisusing IFEFFIT. Physica Scr. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. Iron Oxides in the Laboratory, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Simon, L.; François, M.; Refait, P.; Renaudin, G.; Lelaurain, M.; Génin, J.-M.R. Structure of the Fe(II-III) layered double hydroxysulphate green rust two from Rietveld analysis. Solid State Sci. 2003, 5, 327–334. [Google Scholar] [CrossRef]
- Manceau, A.; Drits, V.A. Local Structure of Ferrihydrite and Feroxyhite by Exafs Spectroscopy. Clay Miner. 2018, 28, 165–184. [Google Scholar] [CrossRef]
- Maillot, F.; Morin, G.; Wang, Y.; Bonnin, D.; Ildefonse, P.; Chaneac, C.; Calas, G. New insight into the structure of nanocrystalline ferrihydrite: EXAFS evidence for tetrahedrally coordinated iron(III). Geochim. Cosmochim. Acta 2011, 75, 2708–2720. [Google Scholar] [CrossRef]
- Papassiopi, N.; Pinakidou, F.; Katsikini, M.; Antipas, G.S.; Christou, C.; Xenidis, A.; Paloura, E.C. A XAFS study of plain and composite iron(III) and chromium(III) hydroxides. Chemosphere 2014, 111, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Charlet, L.; Manceau, A.A. X-ray absorption spectroscopic study of the sorption of Cr(III) at the oxide-water interface. J. Colloid Interface Sci. 1992, 148, 443–458. [Google Scholar] [CrossRef]
- Frommer, J.; Nachtegaal, M.; Czekaj, I.; Kretzschmar, R. The Cr X-ray absorption K-edge structure of poorly crystalline Fe(III)-Cr(III)-oxyhydroxides. Am. Mineral. 2010, 95, 1202–1213. [Google Scholar] [CrossRef]
- Christiansen, B.C.; Balic-Zunic, T.; Petit, P.O.; Frandsen, C.; Mørup, S.; Geckeis, H.; Katerinopoulou, A.; Stipp, S.L.S. Composition and structure of an iron-bearing, layered double hydroxide (LDH)—Green rust sodium sulphate. Geochim. Cosmochim. Acta 2009, 73, 3579–3592. [Google Scholar] [CrossRef]
- Pedersen, H.D.; Postma, D.; Jakobsen, R.; Larsen, O. Fast transformation of iron oxyhydroxides by the catalytic action of aqueous Fe(II). Geochim. Cosmochim. Acta 2005, 69, 3967–3977. [Google Scholar] [CrossRef]
- Buerge, I.J.; Hug, S.J. Kinetics and pH Dependence of Chromium(VI) Reduction by Iron(II). Environ. Sci. Technol. 1997, 31, 1426–1432. [Google Scholar] [CrossRef]
- Sedlak, D.L.; Chan, P.G. Reduction of hexavalent chromium by ferrous iron. Geochim. Cosmochim. Acta 1997, 61, 2185–2192. [Google Scholar] [CrossRef]
- Jansen, E.; Kyek, A.; Schäfer, W.; Schwertmann, U. The structure of six-line ferrihydrite. Appl. Phys. A Mater. Sci. Process. 2002, 74, s1004–s1006. [Google Scholar] [CrossRef]
- Michel, F.M.; Ehm, L.; Antao, S.M.; Lee, P.L.; Chupas, P.J.; Liu, G.; Strongin, D.R.; Schoonen, M.A.; Phillips, B.L.; Parise, J.B. The structure of ferrihydrite, a nanocrystalline material. Science 2007, 316, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.C.B.; Bender Koch, C. Reduction of nitrate to ammonium by sulphate green rust: Activation energy and reaction mechanism. Clay Miner. 1998, 33, 87–101. [Google Scholar] [CrossRef]
- Antony, H.; Legrand, L.; Chaussé, A. Carbonate and sulphate green rusts—Mechanisms of oxidation and reduction. Electrochim. Acta 2008, 53, 7146–7156. [Google Scholar] [CrossRef]
- Schwertmann, U.; Gasser, U.; Sticher, H. Chromium-for-iron substitution in synthetic goethites. Geochim. Cosmochim. Acta 1989, 53, 1293–1297. [Google Scholar] [CrossRef]
- Schrupp, D.; Sing, M.; Tsunekawa, M.; Fujiwara, H.; Kasai, S.; Sekiyama, A.; Suga, S.; Muro, T.; Brabers, V.A.M.; Claessen, R. High-energy photoemission on Fe3O4: Small polaron physics and the Verwey transition. Europhys. Lett. 2005, 70, 789–795. [Google Scholar] [CrossRef]
- Katz, J.E.; Zhang, X.; Attenkofer, K.; Chapman, K.W.; Frandsen, C.; Zarzycki, P.; Rosso, K.M.; Falcone, R.W.; Waychunas, G.A.; Gilbert, B. Electron small polarons and their mobility in iron (oxyhydr)oxide nanoparticles. Science 2012, 337, 1200–1203. [Google Scholar] [CrossRef] [PubMed]
- Wander, M.C.F.; Rosso, K.M.; Schoonen, M.A.A. Structure and Charge Hopping Dynamics in Green Rust. J. Phys. Chem. C 2007, 111, 11414–11423. [Google Scholar] [CrossRef]
- Yao, K.; Taniguchi, M.; Nakata, M.; Takahashi, M.; Yamagishi, A. Electrochemical Scanning Tunneling Microscopy Observation of Ordered Surface Layers on an Anionic Clay-Modified Electrode. Langmuir 1998, 14, 2890–2895. [Google Scholar] [CrossRef]
- Hansen, H.C.B. Environmental chemistry of iron (II)–iron (III) LDHs (green rusts). In Layered Double Hydroxides: Present and Future; Rives, V., Ed.; Nova Science Publishers Inc.: New York, NY, USA, 2001; pp. 413–434. [Google Scholar]
- Garg, A.; Matijevic, E. Preparation and properties of uniformly coated inorganic colloidal particles. 2. Chromium hydrous oxide on hematite. Langmuir 1988, 4, 38–44. [Google Scholar] [CrossRef]
- Kendelewicz, T.; Liu, P.; Doyle, C.S.; Brown, G.E. Spectroscopic study of the reaction of aqueous Cr(VI) with Fe3O4 (111) surfaces. Surf. Sci. 2000, 469, 144–163. [Google Scholar] [CrossRef]
- Lai, K.C.K.; Lo, I.M.C. Removal of chromium (VI) by acid-washed zero-valent iron under various groundwater geochemistry conditions. Environ. Sci. Technol. 2008, 42, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Steefel, C.I.; Marcus, M.A.; Bargar, J.R. Kinetics of Fe(II)-catalyzed transformation of 6-line ferrihydrite under anaerobic flow conditions. Environ. Sci. Technol. 2010, 44, 5469–5475. [Google Scholar] [CrossRef] [PubMed]
- Boland, D.D.; Collins, R.N.; Miller, C.J.; Glover, C.J.; Waite, T.D. Effect of solution and solid-phase conditions on the Fe(II)-accelerated transformation of ferrihydrite to lepidocrocite and goethite. Environ. Sci. Technol. 2014, 48, 5477–5485. [Google Scholar] [CrossRef] [PubMed]
- Papassiopi, N.; Vaxevanidou, K.; Christou, C.; Karagianni, E.; Antipas, G.S. Synthesis, characterization and stability of Cr(III) and Fe(III) hydroxides. J. Hazard. Mater. 2014, 264, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Landrot, G.; Ginder-Vogel, M.; Livi, K.; Fitts, J.P.; Sparks, D.L. Chromium(III) oxidation by three poorly-crystalline manganese(IV) oxides. 1. Chromium(III)-oxidizing capacity. Environ. Sci. Technol. 2012, 46, 11594–11600. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.; James, B. Behavior of chromium in soils: III. Oxidation 1. J. Environ. Qual. 1979, 8, 31–35. [Google Scholar] [CrossRef]
- Fredrickson, J.K.; Zachara, J.M.; Kennedy, D.W.; Dong, H.; Onstott, T.C.; Hinman, N.W.; Li, S.-M. Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium. Geochim. Cosmochim. Acta 1998, 62, 3239–3257. [Google Scholar] [CrossRef]
- Ona-Nguema, G.; Abdelmoula, M.; Jorand, F.; Benali, O.; Block, J.-C.; Génin, J.-M.R. Iron(II,III) Hydroxycarbonate Green Rust Formation and Stabilization from Lepidocrocite Bioreduction. Environ. Sci. Technol. 2002, 36, 16–20. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution | Volume (mL) | [CrO42−] (mg L−1) | Green Rust Suspension Density (mg L−1) |
|---|---|---|---|
| C1 | 50 | 69 | 600 |
| C2 | 100 | 35 | 300 |
| C3 | 250 | 14 | 120 |
| C4 | 500 | 6.9 | 60 |
| C5 | 1000 | 3.5 | 30 |
| C6 | 2000 | 1.7 | 15 |
| Weights | |||||||
|---|---|---|---|---|---|---|---|
| Sample | Feroxyhyte | Goethite | Lepidocrocite | Ferrihydrite | Sum | R-Factor | Reduced χ2 |
| C1 24 h | 0.813 | 0.112 | 0.064 | 0.073 | 1.063 | 0.024 | 0.122 |
| C1 7 d | 0.902 | 0.139 | 0 | 0 | 1.041 | 0.0105 | 0.0476 |
| C2 24 h | 0.741 | 0.229 | 0.032 | 0 | 1.002 | 0.01 | 0.0472 |
| C2 7 d | 0.909 | 0.115 | 0.016 | 0.003 | 1.0 | 0.0114 | 0.0521 |
| C3 24 h | 0.746 | 0.243 | 0.027 | 0 | 1.016 | 0.0090 | 0.0435 |
| C3 7 d | 0.874 | 0.128 | 0.028 | 0 | 1.03 | 0.00656 | 0.0297 |
| C4 24 h | 0.794 | 0.181 | 0.047 | 0 | 1.022 | 0.00748 | 0.0355 |
| C4 7 d | 0.906 | 0.085 | 0.052 | 0.002 | 1.044 | 0.0099 | 0.046 |
| C5 24 h | 0.827 | 0.075 | 0.104 | 0.02 | 1.027 | 0.00513 | 0.0242 |
| C5 7 d | 0.809 | 0.075 | 0.096 | 0.059 | 1.039 | 0.00631 | 0.0303 |
| C6 24 h | 0.531 | 0.39 | 0.033 | 0 | 0.953 | 0.019 | 0.0934 |
| C6 7 d | 0.824 | 0.075 | 0.12 | 0.001 | 1.02 | 0.00452 | 0.0214 |
| Sample | Cr-O | Edge: | Cr-Cr | Single Corner: | Cr-Cr | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | R (Å) | σ2 (Å2) | N | R (Å) | σ2 (Å2) § | N ± 50% | R (Å) | σ2 (Å2) § | ||
| C1 7 d | 6 * | 1.97 | 0.0021 ± 0.0006 | 3 * | 3.06 | 0.011 ± 0.0028 | 2.53 | 3.92 | 0.011 ± 0.0028 | 1.97 |
| C2 7 d | 6 * | 1.96 | 0.0023 ± 0.0006 | 3 * | 3.05 | 0.011 ± 0.0026 | 2.79 | 3.92 | 0.011 ± 0.0026 | 1.87 |
| C3 7 d | 6 * | 1.96 | 0.0019 ± 0.0007 | 3 * | 3.04 | 0.011 ± 0.0032 | 2.64 | 3.92 | 0.011 ± 0.0032 | 2.17 |
| C4 7 d | 6 * | 1.97 | 0.0027 ± 0.0006 | 3 * | 3.04 | 0.0104 ± 0.0023 | 1.84 | 3.89 | 0.0104 ± 0.0023 | 1.95 |
| C5 7 d | 6 * | 1.97 | 0.0022 ± 0.0006 | 3 * | 3.03 | 0.01 ± 0.0022 | 1.76 | 3.89 | 0.01 ± 0.0022 | 1.92 |
| C6 7 d | 6 * | 1.97 | 0.0026 ± 0.0006 | 3 * | 3.03 | 0.01 ± 0.0023 | 1.79 | 3.88 | 0.01 ± 0.0023 | 2.15 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
N. Thomas, A.; Eiche, E.; Göttlicher, J.; Steininger, R.; G. Benning, L.; M. Freeman, H.; Dideriksen, K.; Neumann, T. Products of Hexavalent Chromium Reduction by Green Rust Sodium Sulfate and Associated Reaction Mechanisms. Soil Syst. 2018, 2, 58. https://doi.org/10.3390/soilsystems2040058
N. Thomas A, Eiche E, Göttlicher J, Steininger R, G. Benning L, M. Freeman H, Dideriksen K, Neumann T. Products of Hexavalent Chromium Reduction by Green Rust Sodium Sulfate and Associated Reaction Mechanisms. Soil Systems. 2018; 2(4):58. https://doi.org/10.3390/soilsystems2040058
Chicago/Turabian StyleN. Thomas, Andrew, Elisabeth Eiche, Jörg Göttlicher, Ralph Steininger, Liane G. Benning, Helen M. Freeman, Knud Dideriksen, and Thomas Neumann. 2018. "Products of Hexavalent Chromium Reduction by Green Rust Sodium Sulfate and Associated Reaction Mechanisms" Soil Systems 2, no. 4: 58. https://doi.org/10.3390/soilsystems2040058
APA StyleN. Thomas, A., Eiche, E., Göttlicher, J., Steininger, R., G. Benning, L., M. Freeman, H., Dideriksen, K., & Neumann, T. (2018). Products of Hexavalent Chromium Reduction by Green Rust Sodium Sulfate and Associated Reaction Mechanisms. Soil Systems, 2(4), 58. https://doi.org/10.3390/soilsystems2040058