Arsenite Depletion by Manganese Oxides: A Case Study on the Limitations of Observed First Order Rate Constants


Abstract
:1. Introduction
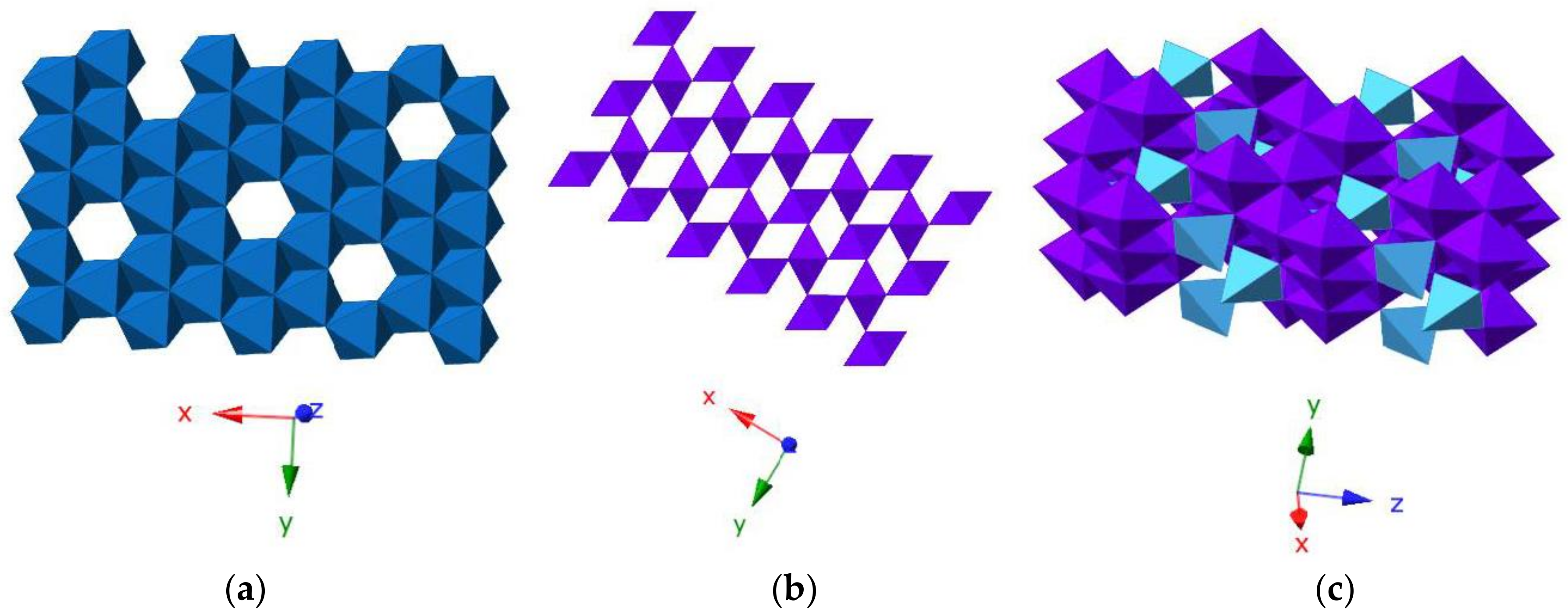
2. Mn Oxide Structure
3. AsIII Depletion Rate Law
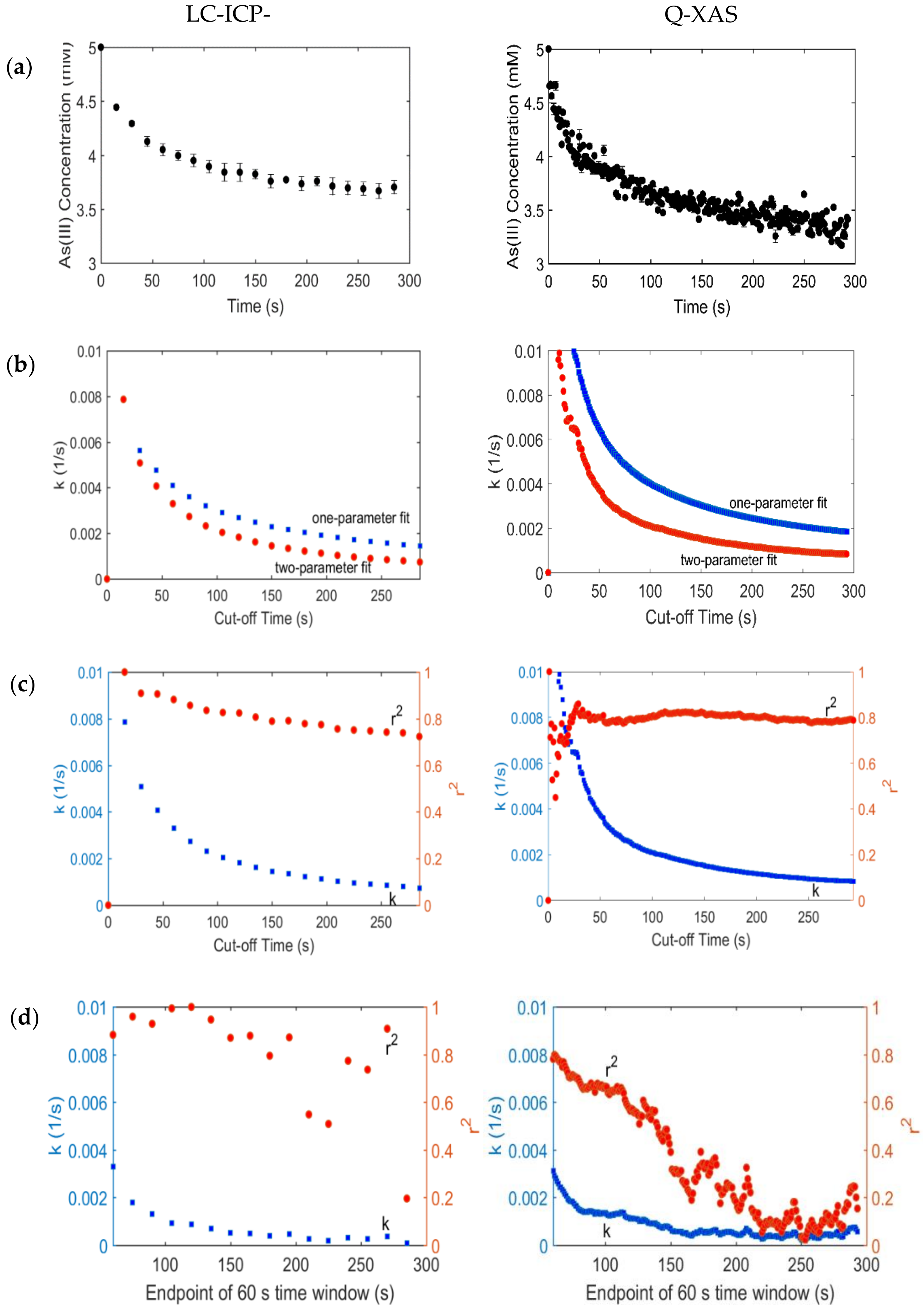
4. Data Analysis
5. Reaction Mechanism
6. Environmental Factors Affecting As Oxidation by Mn Oxides
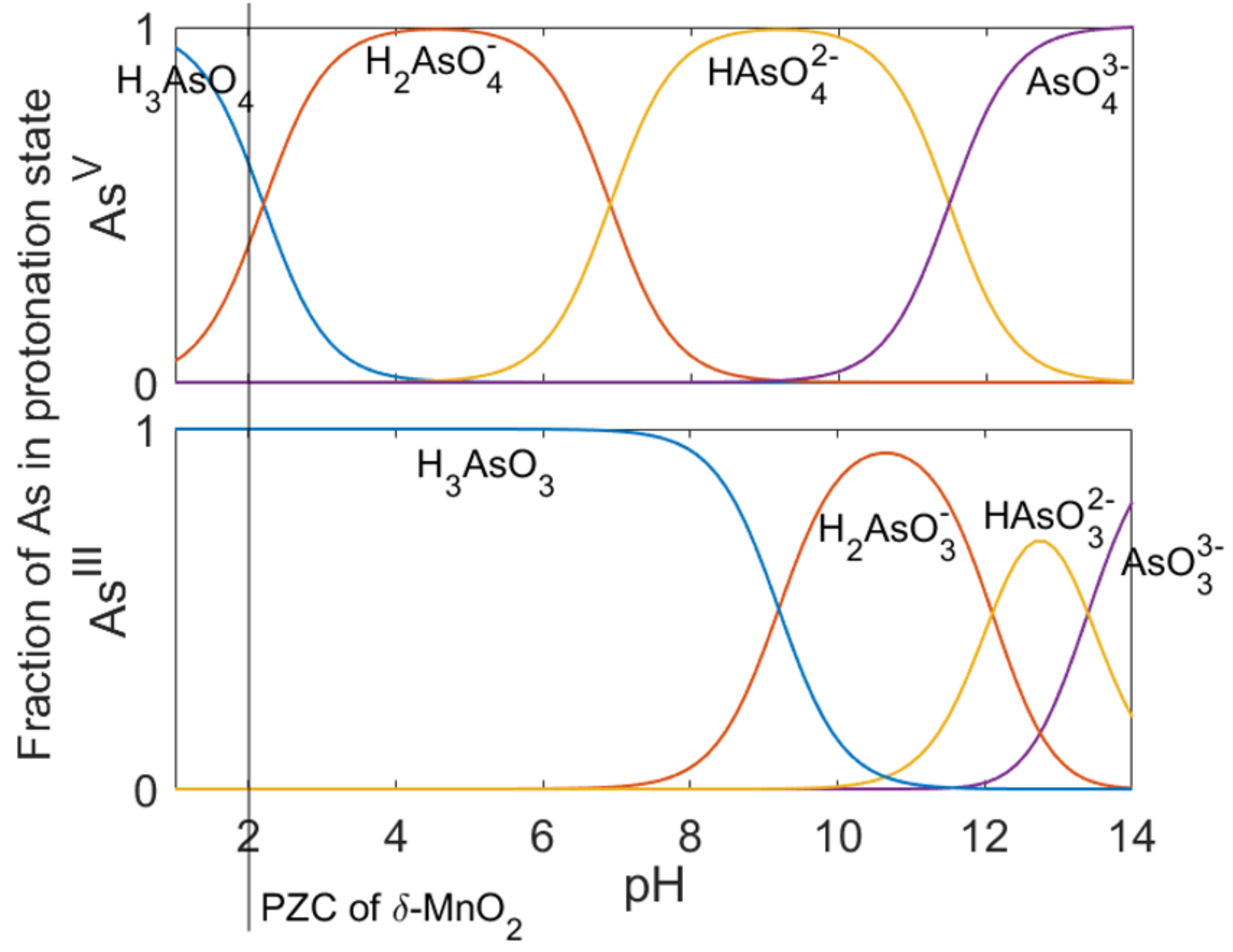
6.1. pH
6.2. Temperature
6.3. Other Oxides
6.4. Competitive Ions
6.5. Other Factors
7. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Ravenscroft, P.; Brammer, H.; Richards, K. Arsenic Solution: A Global Synthesis; Wiley-Blackwell: Hoboken, NJ, USA, 2009; ISBN 978-1-4051-8601-8. [Google Scholar]
- Hem, J.D. Redox processes at surfaces of manganese oxide and their effects on aqueous metal ions. Chem. Geol. 1978, 21, 199–218. [Google Scholar] [CrossRef]
- Borch, T.; Kretzschmar, R.; Kappler, A.; van Cappellen, P.; Ginder-Vogel, M.; Voegelin, A.; Campbell, K. Biogeochemical Redox Processes and their Impact on Contaminant Dynamics. Environ. Sci. Technol. 2010, 44, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Remucal, C.K.; Ginder-Vogel, M. A critical review of the reactivity of manganese oxides with organic contaminants. Environ. Sci. Process. Impacts 2014, 16, 1247–1266. [Google Scholar] [CrossRef] [PubMed]
- Post, J.E. Manganese oxide minerals: Crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. USA 1999, 96, 3447–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nealson, K.H.; Tebo, B.M.; Rosson, R.A. Occurrence and Mechanisms of Microbial Oxidation of Manganese; Academic Press: Cambridge, MA, USA, 1988; Volume 33. [Google Scholar]
- Young, L.B.; Harvey, H.H. The Relative Importance of Manganese and Iron-Oxides and Organic-Matter in the Sorption of Trace-Metals by Surficial Lake-Sediments. Geochim. Cosmochim. Acta 1992, 56, 1175–1186. [Google Scholar] [CrossRef]
- Toner, B.; Manceau, A.; Webb, S.M.; Sposito, G. Zinc sorption to biogenic hexagonal-birnessite particles within a hydrated bacterial biofilm. Geochim. Cosmochim. Acta 2006, 70, 27–43. [Google Scholar] [CrossRef]
- Pang, S.C.; Chin, S.F.; Anderson, M.A. Redox equilibria of iron oxides in aqueous-based magnetite dispersions: Effect of pH and redox potential. J. Colloid Interface Sci. 2007, 311, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, R.M. The Synthesis of Birnessite, Cryptomelane, and Some Other Oxides and Hydroxides of Manganese. Mineral. Mag. 1971, 38, 493–502. [Google Scholar] [CrossRef]
- McKenzie, R.M. The Manganese Oxides in Soils—A Review. J. Plant Nutr. Soil Sci. 1972, 131, 221–242. [Google Scholar] [CrossRef]
- Balistrieri, L.S.; Chao, T.T. Adsorption of selenium by amourphous iron oxyhydroxide and manganese dioxide. Geochem. Cosmochim. Acta 1990, 54, 739–751. [Google Scholar] [CrossRef]
- Scott, M.J.; Morgan, J.J. Reactions at oxide surfaces. 2. Oxidation of Se(IV) by synthetic birnessite. Environ. Sci. Technol. 1996, 30, 1990–1996. [Google Scholar] [CrossRef]
- Foster, A.L.; Gordon, E.; Brown, J.; Parks, G.A. X-ray absorption fine structure study of As (V) and Se (IV) sorption complexes on hydrous Mn oxides. Geochim. Cosmochim. Acta 2003, 67, 1937–1953. [Google Scholar] [CrossRef]
- Eary, L.E.; Rai, D. Kinetics of chromium (III) oxidation to chromium (VI) by reaction with manganese dioxide. Environ. Sci. Technol. 1987, 21, 1187–1193. [Google Scholar] [CrossRef]
- Landrot, G.; Ginder-Vogel, M.; Sparks, D.L. Kinetics of chromium(III) oxidation by manganese(IV) oxides using quick scanning X-ray absorption fine structure spectroscopy (Q-XAFS). Environ. Sci. Technol. 2010, 44, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.M.; Fuller, C.C.; Tebo, B.M.; Bargar, J.R. Determination of uranyl incorporation into biogenic manganese oxides using X-ray absorption spectroscopy and scattering. Environ. Sci. Technol. 2006, 40, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lee, S.-W.; Kapoor, P.; Tebo, B.M.; Giammar, D.E. Uraninite oxidation and dissolution induced by manganese oxide: A redox reaction between two insoluble minerals. Geochim. Cosmochim. Acta 2013, 100, 24–40. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef]
- Ahmad, S.A.; Sayed, M.H.S.; Khan, M.H.; Karim, M.N.; Haque, M.A.; Bhuiyan, M.S.A.; Rahman, M.S.; Faruquee, M.H. Sociocultural aspects of arsenicosis in Bangladesh: Community perspective. J. Environ. Sci. Health Part A 2007, 42, 1945–1958. [Google Scholar] [CrossRef] [PubMed]
- Argos, M.; Kalra, T.; Rathouz, P.J.; Pierce, B.; Ahsan, H.; Slavkovich, V.M.; Graziano, J.; Islam, T.; Ahmed, A.; Rakibuz-Zaman, M.; et al. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): A prospective cohort study. Lancet 2010, 376, 252–258. [Google Scholar] [CrossRef]
- Ahmed, K.M.; Bhattacharya, P.; Hasan, M.A.; Akhter, S.H.; Alam, S.M.M.; Bhuyian, M.A.H.; Imam, M.B.; Khan, A.A.; Sracek, O. Arsenic enrichment in groundwater of the alluvial aquifers in Bangladesh: An overview. Appl. Geochem. 2004, 19, 181–200. [Google Scholar] [CrossRef]
- Gillispie, E.C.; Andujar, E.; Polizzotto, M.L. Chemical controls on abiotic and biotic release of geogenic arsenic from Pleistocene aquifer sediments to groundwater. Environ. Sci. Process. Impacts 2016, 18, 1090–1103. [Google Scholar] [CrossRef] [PubMed]
- Polizzotto, M.L.; Kocar, B.D.; Benner, S.G.; Sampson, M.; Fendorf, S.E. Near-surface wetland sediments as a source of arsenic release to ground water in Asia. Nature 2008, 454, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.C.; Schaefer, M.V.; Cock-Esteb, A.; Li, J.; Fendorf, S. Depth Stratification Leads to Distinct Zones of Manganese and Arsenic Contaminated Groundwater. Environ. Sci. Technol. 2017, 51, 8926–8932. [Google Scholar] [CrossRef] [PubMed]
- Atsdr, U. Toxicological Profile for Arsenic; Agency for Toxic Substances and Disease Registry, Division of Toxicology: Atlanta, GA, USA, 2007; Volume 24. [Google Scholar] [CrossRef]
- De Meyer, C.M.C.; Rodríguez, J.M.; Carpio, E.A.; García, P.A.; Stengel, C.; Berg, M. Arsenic, manganese and aluminum contamination in groundwater resources of Western Amazonia (Peru). Sci. Total Environ. 2017, 607–608, 1437–1450. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, J.; Berg, M.; Stengel, C.; Sampson, M.L. Arsenic and manganese contamination of drinking water resources in Cambodia: Coincidence of risk areas with low relief topography. Environ. Sci. Technol. 2007, 41, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Sthiannopkao, S.; Kim, K.W.; Sotham, S.; Choup, S. Arsenic and manganese in tube well waters of Prey Veng and Kandal Provinces, Cambodia. Appl. Geochem. 2008, 23, 1086–1093. [Google Scholar] [CrossRef]
- Bacquart, T.; Frisbie, S.; Mitchell, E.; Grigg, L.; Cole, C.; Small, C.; Sarkar, B. Multiple inorganic toxic substances contaminating the groundwater of Myingyan Township, Myanmar: Arsenic, manganese, fluoride, iron, and uranium. Sci. Total Environ. 2015, 517, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Naseem, S.; Mcarthur, J.M. Arsenic and other water-quality issues affecting groundwater, Indus alluvial plain, Pakistan. Hydrol. Process. 2018, 32, 1235–1253. [Google Scholar] [CrossRef]
- Mcarthur, J.M.; Sikdar, P.K.; Leng, M.J.; Ghosal, U.; Sen, I. Groundwater Quality beneath an Asian Megacity on a Delta: Kolkata’s (Calcutta’s) Disappearing Arsenic and Present Manganese. Environ. Sci. Technol. 2018, 52, 5161–5172. [Google Scholar] [CrossRef] [PubMed]
- Gillispie, E.; Polizzotto, M.; Jones, M. Investigating the role of sediment manganese as a potential predictor for future arsenic contamination of groundwater. In Abstracts of Papers of the American Chemical Society; American Chemical Society: Washington, DC, USA, 2017. [Google Scholar]
- Scott, M.J.; Morgan, J.J. Reactions at oxide surfaces. 1. Oxidation of As(III) by synthetic Birnessite. Environ. Sci. Technol. 1995, 29, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, H.W.; Canning, G.W.; Bancroft, G.M. XPS study of reductive dissolution of 7Å-birnessite by H3AsO3, with constraints on reaction mechanism. Geochim. Cosmochim. Acta 1998, 62, 2097–2110. [Google Scholar] [CrossRef]
- Tournassat, C.; Charlet, L.; Bosbach, D.; Manceau, A. Arsenic(III) Oxidation by Birnessite and Precipitation of Manganese(II) Arsenate. Environ. Sci. Technol. 2002, 36, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Lafferty, B.J.; Ginder-Vogel, M.; Sparks, D.L. Arsenite oxidation by a poorly crystalline manganese-oxide 1. Stirred-flow experiments. Environ. Sci. Technol. 2010, 44, 8460–8466. [Google Scholar] [CrossRef] [PubMed]
- Oscarson, D.W.; Huang, P.M.; Liaw, W.K.; Hammer, U.T. Kinetics of oxidation of arsenite by various manganese dioxides. Soil Sci. Soc. Am. J. 1983, 47, 644–648. [Google Scholar] [CrossRef]
- Tebo, B.M.; Bargar, J.R.; Clement, B.G.; Dick, G.J.; Murray, K.J.; Parker, D.; Verity, R.; Webb, S.M. Biogenic Manganese Oxides: Properties and Mechanisms of Formation. Annu. Rev. Earth Planet. Sci. 2004, 32, 287–328. [Google Scholar] [CrossRef]
- Duckworth, O.W.; Rivera, N.A.; Gardner, T.G.; Andrews, M.Y.; Santelli, C.M.; Polizzotto, M.L. Morphology, structure, and metal binding mechanisms of biogenic manganese oxides in a superfund site treatment system. Environ. Sci. Process. Impacts 2017, 19, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Ilton, E.S.; Post, J.E.; Heaney, P.J.; Ling, F.T.; Kerisit, S.N. XPS determination of Mn oxidation states in Mn (hydr)oxides. Appl. Surf. Sci. 2016, 366, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Hem, J.D. Rates of manganese oxidation in aqueous systems. Geochim. Cosmochim. Acta 1981, 45, 1369–1374. [Google Scholar] [CrossRef]
- Villalobos, M.; Toner, B.; Bargar, J.; Sposito, G. Characterization of the manganese oxide produced by Pseudomonas putida strain MnB1. Geochim. Cosmochim. Acta 2003, 67, 2649–2662. [Google Scholar] [CrossRef]
- Webb, S.M.; Dick, G.J.; Bargar, J.R.; Tebo, B.M. Evidence for the presence of Mn(III) intermediates in the bacterial oxidation of Mn(II). Proc. Natl. Acad. Sci. USA 2005, 102, 5558–5563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droz, B.; Dumas, N.; Duckworth, O.W.; Peña, J. A comparison of the sorption reactivity of bacteriogenic and mycogenic Mn oxide nanoparticles. Environ. Sci. Technol. 2015, 49, 4200–4208. [Google Scholar] [CrossRef] [PubMed]
- Santelli, C.M.; Webb, S.M.; Dohnalkova, A.C.; Hansel, C.M. Diversity of Mn oxides produced by Mn(II)-oxidizing fungi. Geochim. Cosmochim. Acta 2011, 75, 2762–2776. [Google Scholar] [CrossRef]
- Zhu, M. Structure and Reactivity Study of Biotic and Abiotic Poorly Crystalline Manganese Oxides. Ph.D. Thesis, University of Delaware, Newark, DE, USA, 2010. [Google Scholar]
- Zhu, M.; Paul, K.W.; Kubicki, J.D.; Sparks, D.L. Quantum chemical study of arsenic(III, V) adsorption on Mn-oxides: Implications for arsenic(III) oxidation. Environ. Sci. Technol. 2009, 43, 6655–6661. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zu, Y.; Tan, W.; Liu, F. Arsenite oxidation by three types of manganese oxides. J. Environ. Sci. 2006, 18, 292–298. [Google Scholar]
- Fischel, M.H.; Fischel, J.S.; Lafferty, B.J.; Sparks, D.L. The influence of environmental conditions on kinetics of arsenite oxidation by manganese-oxides. Geochem. Trans. 2015, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Marafatto, F.F.; Lanson, B.; Peña, J. Crystal growth and aggregation in suspensions of δ-MnO2 nanoparticles: Implications for surface reactivity. Environ. Sci. Nano 2018, 5, 497–508. [Google Scholar] [CrossRef]
- Feng, X.; Tan, W.; Liu, F.; Ruan, H.D.; Jizheng, H. Oxidation of As(III) by several manganese oxide minerals in absence and presence of goethite. Acta Geol. Sin. 2006, 80, 249–256. [Google Scholar]
- Wang, Y.; Benkaddour, S.; Marafatto, F.F.; Peña, J. Diffusion- and pH-Dependent Reactivity of Layer-Type MnO2: Reactions at Particle Edges versus Vacancy Sites. Environ. Sci. Technol. 2018, 52, 3476–3485. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Feng, X.; Villalobos, M.; Tan, W.; Liu, F. Sorption behavior of heavy metals on birnessite: Relationship with its Mn average oxidation state and implications for types of sorption sites. Chem. Geol. 2012, 292–293, 25–34. [Google Scholar] [CrossRef]
- Villalobos, M.; Escobar-Quiroz, I.N.; Salazar-Camacho, C. The influence of particle size and structure on the sorption and oxidation behavior of birnessite: I. Adsorption of As(V) and oxidation of As(III). Geochim. Cosmochim. Acta 2014, 125, 564–581. [Google Scholar] [CrossRef]
- Lafferty, B.J.; Ginder-vogel, M.; Zhu, M.; Livi, K.J.T.; Sparks, D.L. Arsenite oxidation by a poorly crystalline manganese-oxide. 2. Results from X-ray absorption spectroscopy and X-ray diffraction. Environ. Sci. Technol. 2010, 44, 8467–8472. [Google Scholar] [CrossRef] [PubMed]
- Balgooyen, S.; Alaimo, P.J.; Remucal, C.K.; Ginder-Vogel, M. Structural Transformation of MnO2 during the Oxidation of Bisphenol A. Environ. Sci. Technol. 2017, 51, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Zhai, L.M.; Tan, W.F.; Liu, F.; He, J.Z. Adsorption and redox reactions of heavy metals on synthesized Mn oxide minerals. Environ. Pollut. 2007, 147, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Farrow, C.L.; Post, J.E.; Livi, K.J.T.; Billinge, S.J.L.; Ginder-Vogel, M.; Sparks, D.L. Structural study of biotic and abiotic poorly-crystalline manganese oxides using atomic pair distribution function analysis. Geochim. Cosmochim. Acta 2012, 81, 39–55. [Google Scholar] [CrossRef]
- Murray, J.W. The interaction of metal ions at the manganese dioxide solution interface. Geochim. Cosmochim. Acta 1975, 39, 505–519. [Google Scholar] [CrossRef]
- Händel, M.; Rennert, T.; Totsche, K.U. A simple method to synthesize birnessite at ambient pressure and temperature. Geoderma 2013, 193–194, 117–121. [Google Scholar] [CrossRef]
- Zhu, M.; Ginder-Vogel, M.; Parikh, S.J.; Feng, X.H.; Sparks, D.L. Cation effects on the layer structure of biogenic Mn-oxides. Environ. Sci. Technol. 2010, 44, 4465–4471. [Google Scholar] [CrossRef] [PubMed]
- Ginder-Vogel, M.; Landrot, G.; Fischel, J.S.; Sparks, D.L. Quantification of rapid environmental redox processes with quick-scanning X-ray absorption spectroscopy (Q-XAS). Proc. Natl. Acad. Sci. USA 2009, 106, 16124–16128. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, Y.L.; Gu, J.D. Oxidation of As(III) by MnO2 in the absence and presence of Fe(II) under acidic conditions. Geochim. Cosmochim. Acta 2011, 75, 368–379. [Google Scholar] [CrossRef]
- Fischel, J.S.; Fischel, M.H.; Sparks, D.L. Advances in understanding reactivity of manganese oxides with arsenic and chromium in environmental systems. In Advances in the Environmental Biogeochemistry of Manganese Oxides; American Chemical Society: Washington, DC, USA, 2015; Volume 1197, pp. 1–27. [Google Scholar] [CrossRef]
- Jones, L.C.; Lafferty, B.J.; Sparks, D.L. Additive and competitive effects of bacteria and Mn oxides on arsenite oxidation kinetics. Environ. Sci. Technol. 2012, 46, 6548–6555. [Google Scholar] [CrossRef] [PubMed]
- Power, L.E.; Arai, Y.; Sparks, D.L. Zinc adsorption effects on arsenite oxidation kinetics at the birnessite-water interface. Environ. Sci. Technol. 2005, 39, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Katsoyiannis, I.A.; Zouboulis, A.I.; Jekel, M. Kinetics of Bacterial As(III) Oxidation and Subsequent As(V) Removal by Sorption onto Biogenic Manganese Oxides during Groundwater Treatment. Ind. Eng. Chem. Res. 2003, 43, 486–493. [Google Scholar] [CrossRef]
- Tani, Y.; Miyata, N.; Ohashi, M.; Ohnuki, T.; Seyama, H.; Iwahori, K.; Soma, M. Interaction of inorganic arsenic with biogenic manganese oxide produced by a Mn-oxidizing fungus, strain KR21-2. Environ. Sci. Technol. 2004, 38, 6618–6624. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.; Fendorf, S.E.; Bostick, B.; Suarez, D.L. Arsenic(III) Oxidation and Arsenic(V) Adsorption Reactions on Synthetic Birnessite. Environ. Sci. Technol. 2002, 36, 976–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.N.; Walker, J.R.; Hayes, T.H. Reaction scheme for the oxidation of As(III) to As(V) by birnessite. Clays Clay Miner. 1990, 38, 549–555. [Google Scholar] [CrossRef]
- Hou, J.; Xiang, Y.; Zheng, D.; Li, Y.; Xue, S.; Wu, C.; Hartley, W.; Tan, W. Morphology-dependent enhancement of arsenite oxidation to arsenate on birnessite-type manganese oxide. Chem. Eng. J. 2017, 327, 235–243. [Google Scholar] [CrossRef]
- Thanabalasingam, P.; Pickering, W.F. Effect of pH on interaction between As(III) or As(V) and manganese(IV) oxide. Water Air Soil Pollut. 1986, 29, 205–216. [Google Scholar] [CrossRef]
- Parikh, S.J.; Lafferty, B.J.; Meade, T.G.; Sparks, D.L. Evaluating environmental influences on As(III) oxidation kinetics by a poorly crystalline Mn-oxide. Environ. Sci. Technol. 2010, 44, 3772–3778. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.F. Pseudo first-order kinetics. J. Chem. Educ. 1972, 49, 663. [Google Scholar] [CrossRef]
- Cern, M. Krautite, Mn (HTOXAsOTOH): Crystal structure, hydrogen bonding and relations with haidingerite and pharmacolite. Am. Mineral. 1979, 64, 1248–1254. [Google Scholar]
- Driehaus, W.; Seith, R.; Jekel, M. Oxidation of arsenate(III) with manganese oxides in water treatment. Water Res. 1995, 29, 297–305. [Google Scholar] [CrossRef]
- Mitsunobu, S.; Takahashi, Y.; Uruga, T. Observation of chemical reactions at the solid-water interface by quick XAFS combined with a column reactor. Anal. Chem. 2006, 78, 7040–7043. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.V.; Porkodi, K.; Rocha, F. Langmuir-Hinshelwood kinetics—A theoretical study. Catal. Commun. 2008, 9, 82–84. [Google Scholar] [CrossRef]
- Silvester, E.; Manceau, A.; Drits, V.A. Structure of synthetic monoclinic Na-rich birnessite and hexagonal birnessite: II. Results from chemical studies and EXAFS spectroscopy. Am. Mineral. 1997, 82, 962–978. [Google Scholar] [CrossRef]
- Birkner, N.; Navrotsky, A. Thermodynamics of manganese oxides: Sodium, potassium, and calcium birnessite and cryptomelane. Proc. Natl. Acad. Sci. USA 2017, 114, E1046–E1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Feng, X.; Tan, W.; Liu, F.; Ding, S. Relation of lead adsorption on birnessites with different average oxidation states of manganese and release of Mn2+/H+/K+. J. Environ. Sci. 2009, 21, 520–526. [Google Scholar] [CrossRef]
- Institut International du Manganèse (IMnI). Manganese in Groundwater: Research and Potential Risks; Institut International du Manganèse: Paris, France, 2013; pp. 2–3. [Google Scholar]
- Perez-Benito, J.F. Reduction of colloidal manganese dioxide by manganese(II). J. Colloid Interface Sci. 2002, 248, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.J. Manganese in natural waters and Earth’s crust: Its availability to organisms. In Metal Ions in Biological Systems, Manganese and Its Role in Biological Processes; Sigel, A., Sigel, H., Eds.; Metal Ions in Biological Systems; Marcel Dekker: New York, NY, USA, 2000; pp. 1–33. ISBN 0824702883. [Google Scholar]
- Elzinga, E.J.; Kustka, A.B. A Mn-54 radiotracer study of Mn isotope solid-liquid exchange during reductive transformation of vernadite (δ-MnO2) by aqueous Mn(II). Environ. Sci. Technol. 2015, 49, 4310–4316. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.J. Kinetics of Adsorption and Redox Processes on Iron and Manganese Oxides: Reactions of As(III) and Se(IV) at Goethite and Birnessite Surfaces. Ph.D. Thesis, California Institute of Technology, Pasadena, CA, USA, 1991. [Google Scholar]
- Churchman, G.J.; Lowe, D.J. Handbook of Soil Sciences: Properties and Processes; Huang, P.M., Li, Y., Sumner, M.E., Eds.; CRC Press: Boca Raton, FL, USA, 2012; ISBN 978-1-4398-0305-9. [Google Scholar]
- Villalobos, M. The Role of Surface Edge Sites in Metal(loid) Sorption to Poorly-Crystalline Birnessites. In Advances in the Environmental Biogeochemistry of Manganese Oxides; American Chemical Society: Washington, DC, USA, 2015; pp. 65–87. [Google Scholar]
- Fendorf, S.; Nico, P.S.; Kocar, B.D.; Masue, Y.; Tufano, K.J. Arsenic Chemistry in Soils and Sediments. In Developments in Soil Science; Elsevier: New York, NY, USA, 2010; Volume 34, pp. 357–378. ISBN 0166-2481. [Google Scholar]
- Machesky, M.L. Influence of temperature on ion adsorption by hydrous metal oxides. In Chemical Modeling of Aqueous Systems II; ACS: Washington, DC, USA, 1990; Volume 416, pp. 282–292. [Google Scholar] [CrossRef]
- Kosmulski, M. Isoelectric points and points of zero charge of metal (hydr)oxides: 50 years after Parks’ review. Adv. Colloid Interface Sci. 2016, 238, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Kosmulski, M. Surface Charging and Points of Zero Charge; Surfactant Science; CRC Press: Boca Raton, FL, USA, 2009; Volume 145, ISBN 9786612111105. [Google Scholar]
- Xu, H.; Allard, B.; Grimvall, A. Influence of pH and organic substance on the adsorption of As(V) on geologic materials. Water Air Soil Pollut. 1988, 40, 293–305. [Google Scholar] [CrossRef]
- Bowell, R.J. Sorption of arsenic by iron oxides and oxyhydroxides in soils. Appl. Geochem. 1994, 9, 279–286. [Google Scholar] [CrossRef]
- Welch, A.H.; Stollenwerk, K.G. (Eds.) Arsenic in Ground Water: Geochemistry and Occurance; Springer Science & Business Media: Berlin, Germany, 2003. [Google Scholar]
- Liu, H.; Chen, T.; Frost, R.L. An overview of the role of goethite surfaces in the environment. Chemosphere 2014, 103, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmulski, M.; Maczka, E.; Jartych, E.; Rosenholm, J.B. Synthesis and characterization of goethite and goethite–hematite composite: Experimental study and literature survey. Adv. Colloid Interface Sci. 2003, 103, 57–76. [Google Scholar] [CrossRef]
- Gupta, S.K.; Chen, K.Y. Arsenic Removal by Adsorption. J. Water Pollut. Control Feder. 1978, 50, 493–506. [Google Scholar]
- Masue, Y.; Loeppert, R.H.; Kramer, T.A. Arsenate and arsenite adsorption and desorption behavior on coprecipitated aluminum: Iron hydroxides. Environ. Sci. Technol. 2007, 41, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Waychunas, G.A.; Rea, B.A.; Fuller, C.C.; Davis, J.A. Surface chemistry of ferrihydrite: Part 1. EXAFS studies ongeometrie of coprecipitated and adsorbed arsenate. Geochim. Cosmochim. Acta 1993, 57, 2251–2269. [Google Scholar] [CrossRef]
- Sun, X.; Doner, H.E. An investigation of arsenate and arsenite bonding structures on goethite by FTIR. Soil Sci. 1996, 161, 865–872. [Google Scholar] [CrossRef]
- Sun, X.; Doner, H.E. Adsorption and oxidation of arsenite on goethite. Soil Sci. 1998, 163, 278–287. [Google Scholar] [CrossRef]
- Goldberg, S.; Johnston, C.T. Mechanisms of Arsenic Adsorption on Amorphous Oxides Evaluated Using Macroscopic Measurements, Vibrational Spectroscopy, and Surface Complexation Modeling. J. Colloid Interface Sci. 2001, 234, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Ona-Nguema, G.; Morin, G.; Juillot, F.; Calas, G.; Brown, G.E. EXAFS analysis of arsenite adsorption onto two-line ferrihydrite, hematite, goethite, and lepidocrocite. Environ. Sci. Technol. 2005, 39, 9147–9155. [Google Scholar] [CrossRef] [PubMed]
- Catalano, J.G.; Zhang, Z.; Park, C.; Fenter, P.; Bedzyk, M.J. Bridging arsenate surface complexes on the hematite (0 1 2) surface. Geochim. Cosmochim. Acta 2007, 71, 1883–1897. [Google Scholar] [CrossRef]
- Manning, B.A.; Fendorf, S.E.; Goldberg, S. Surface structures and stability of arsenic(III) on goethite: Spectroscopic evidence for inner-sphere complexes. Environ. Sci. Technol. 1998, 32, 2383–2388. [Google Scholar] [CrossRef]
- Kocar, B.D.; Herbel, M.J.; Tufano, K.J.; Fendorf, S. Contrasting effects of dissimilatory iron(III) and arsenic(V) reduction on arsenic retention and transport. Environ. Sci. Technol. 2006, 40, 6715–6721. [Google Scholar] [CrossRef] [PubMed]
- Catalano, J.G.; Park, C.; Fenter, P.; Zhang, Z. Simultaneous inner- and outer-sphere arsenate adsorption on corundum and hematite. Geochim. Cosmochim. Acta 2008, 72, 1986–2004. [Google Scholar] [CrossRef]
- Wang, Y.; Morin, G.; Ona-Nguema, G.; Menguy, N.; Juillot, F.; Aubry, E.; Guyot, F.; Calas, G.; Brown, G.E. Arsenite sorption at the magnetite-water interface during aqueous precipitation of magnetite: EXAFS evidence for a new arsenite surface complex. Geochim. Cosmochim. Acta 2008, 72, 2573–2586. [Google Scholar] [CrossRef]
- Raven, K.P.; Jain, A.; Loeppert, R.H. Arsenite and arsenate adsorption on ferrihydrite: Kinetics, equilibrium, and adsorption envelopes. Environ. Sci. Technol. 1998, 32, 344–349. [Google Scholar] [CrossRef]
- Dixit, S.; Hering, J.G. Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: Implications for arsenic mobility. Environ. Sci. Technol. 2003, 37, 4182–4189. [Google Scholar] [CrossRef] [PubMed]
- Herbel, M.; Fendorf, S. Biogeochemical processes controlling the speciation and transport of arsenic within iron coated sands. Chem. Geol. 2006, 228, 16–32. [Google Scholar] [CrossRef]
- Ying, S.C.; Kocar, B.D.; Fendorf, S.E. Oxidation and competitive retention of arsenic between iron- and manganese oxides. Geochim. Cosmochim. Acta 2012, 96, 294–303. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, H.; Qu, J.; Jefferson, W. Arsenate uptake and arsenite simultaneous sorption and oxidation by Fe-Mn binary oxides: Influence of Mn/Fe ratio, pH, Ca2+, and humic acid. J. Colloid Interface Sci. 2012, 366, 141–146. [Google Scholar] [CrossRef] [PubMed]
- He, Y.T.; Hering, J.G. Enhancement of Arsenic(III) Sequestration by Manganese Oxides in the Presence of Iron(II). Water Air Soil Pollut. 2009, 203, 359–368. [Google Scholar] [CrossRef]
- Tufano, K.J.; Fendorf, S. Confounding impacts of iron reduction on arsenic retention. Environ. Sci. Technol. 2008, 42, 4777–4783. [Google Scholar] [CrossRef] [PubMed]
- Ehlert, K.; Mikutta, C.; Kretzschmar, R. Effects of manganese oxide on arsenic reduction and leaching from contaminated floodplain soil. Environ. Sci. Technol. 2016, 50, 9251–9261. [Google Scholar] [CrossRef] [PubMed]
- Oscarson, D.W.; Huang, P.M.; Hammer, U.T.; Liaw, W.K. Oxidation and Sorption of Arsenite by Manganese-Dioxide As Influenced By Surface-Coatings of Iron and Aluminum-Oxides and Calcium-Carbonate. Water Air Soil Pollut. 1983, 20, 233–244. [Google Scholar] [CrossRef]
- Deschamps, E.; Ciminelli, V.S.T.; Weidler, P.G.; Ramos, A.Y. Arsenic sorption onto soils enriched in Mn and Fe minerals. Clays Clay Miner. 2003, 51, 197–204. [Google Scholar] [CrossRef]
- Deschamps, E.; Ciminelli, V.S.T.; Höll, W.H. Removal of As(III) and As(V) from water using a natural Fe and Mn enriched sample. Water Res. 2005, 39, 5212–5220. [Google Scholar] [CrossRef] [PubMed]
- Ouvrard, S.; de Donato, P.; Simonnot, M.O.; Begin, S.; Ghanbaja, J.; Alnot, M.; Duval, Y.B.; Lhote, F.; Barres, O.; Sardin, M. Natural manganese oxide: Combined analytical approach for solid characterization and arsenic retention. Geochim. Cosmochim. Acta 2005, 69, 2715–2724. [Google Scholar] [CrossRef]
- Zhang, G.S.; Qu, J.H.; Liu, H.J.; Liu, R.P.; Li, G.T. Removal mechanism of As(III) by a novel Fe-Mn binary oxide adsorbent: Oxidation and sorption. Environ. Sci. Technol. 2007, 41, 4613–4619. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Tong, M. Efficient removal of trace arsenite through oxidation and adsorption by magnetic nanoparticles modified with Fe-Mn binary oxide. Water Res. 2013, 47, 3411–3421. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhang, G.; Li, H. Efficient removal of arsenic from water using a granular adsorbent: Fe-Mn binary oxide impregnated chitosan bead. Bioresour. Technol. 2015, 193, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.P.; Zhang, G.; Jefferson, W.A.; Perfilev, A.V.; Liu, R.; Liu, H.; Qu, J. Adsorption of aromatic organoarsenic compounds by ferric and manganese binary oxide and description of the associated mechanism. Chem. Eng. J. 2017, 309, 577–587. [Google Scholar] [CrossRef]
- Behrendt, H.; Boekhold, A. Phosphorus saturation in soils and groundwaters. Land Degrad. Dev. 1993, 4, 233–243. [Google Scholar] [CrossRef]
- Masue-Slowey, Y.; Kocar, B.D.; Bea Jofré, S.A.; Mayer, K.U.; Fendorf, S. Transport implications resulting from internal redistribution of arsenic and iron within constructed soil aggregates. Environ. Sci. Technol. 2011, 45, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Persson, I. Hydrated metal ions in aqueous solution: How regular are their structures? Pure Appl. Chem. 2010, 82, 1901–1917. [Google Scholar] [CrossRef]
- Tani, Y.; Ohashi, M.; Miyata, N.; Seyama, H.; Iwahori, K.; Soma, M. Sorption of Co.(II), Ni(II), and Zn(II) on biogenic manganese oxides produced by a Mn-oxidizing fungus, strain KR21-2. J. Environ. Sci. Health Part A 2004, 39, 2641–2660. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Rosen, B.P.; Phung, L.T.; Silver, S. Microbial arsenic: From geocycles to genes and enzymes. FEMS Microbiol. Rev. 2002, 26, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Oremland, R.S.; Stolz, J.F. The ecology of arsenic. Science 2003, 300, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Rhine, E.D.; Garcia-Dominguez, E.; Phelps, C.D.; Young, L.Y. Environmental microbes can speciate and cycle arsenic. Environ. Sci. Technol. 2005, 39, 9569–9573. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dominguez, E.; Mumford, A.; Rhine, E.D.; Paschal, A.; Young, L.Y. Novel autotrophic arsenite-oxidizing bacteria isolated from soil and sediments. FEMS Microbiol. Ecol. 2008, 66, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, S.; Phung, L.T. Genes and enzymes involved in bacterial oxidation and reduction of inorganic arsenic. Appl. Environ. Microbiol. 2005, 71, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.C.; Kocar, B.D.; Griffis, S.D.; Fendorf, S. Competitive Microbially and Mn Oxide Mediated Redox Processes Controlling Arsenic Speciation and Partitioning. Environ. Sci. Technol. 2011, 45, 5572–5579. [Google Scholar] [CrossRef] [PubMed]
- Shumlas, S.L.; Singireddy, S.; Thenuwara, A.C.; Attanayake, N.H.; Reeder, R.J.; Strongin, D.R. Oxidation of arsenite to arsenate on birnessite in the presence of light. Geochem. Trans. 2016, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.J. Kinetics of reaction between O2 and Mn(II) species in aqueous solutions. Geochim. Cosmochim. Acta 2005, 69, 35–48. [Google Scholar] [CrossRef]
- Feng, X.; Wang, P.; Shi, Z.; Kwon, K.D.; Zhao, H.; Yin, H.; Lin, Z.; Zhu, M.; Liang, X.; Liu, F.; Sparks, D.L. A Quantitative Model for the Coupled Kinetics of Arsenic Adsorption/Desorption and Oxidation on Manganese Oxides. Environ. Sci. Technol. Lett. 2018, 5, 175–180. [Google Scholar] [CrossRef]
- Sherman, D.M.; Randall, S.R. Surface complexation of arsenic(V) to iron(III) (hydr)oxides: Structural mechanism from ab initio molecular geometries and EXAFS spectroscopy. Geochim. Cosmochim. Acta 2003, 67, 4223–4230. [Google Scholar] [CrossRef]
- Appelo, C.A.J.; Van Der Weiden, M.J.J.; Tournassat, C.; Charlet, L. Surface complexation of ferrous iron and carbonate on ferrihydrite and the mobilization of arsenic. Environ. Sci. Technol. 2002, 36, 3096–3103. [Google Scholar] [CrossRef] [PubMed]
- Sverjensky, D.A.; Fukushi, K. A predictive model (ETLM) for As(III) adsorption and surface speciation on oxides consistent with spectroscopic data. Geochim. Cosmochim. Acta 2006, 70, 3778–3802. [Google Scholar] [CrossRef]
- Appelo, C.A.J.; Postma, D. A consistent model for surface complexation on birnessite(-MnO2) and its application to a column experiment. Geochim. Cosmochim. Acta 1999, 63, 3039–3048. [Google Scholar] [CrossRef]
- Tonkin, J.W.; Balistrieri, L.S.; Murray, J.W. Modeling sorption of divalent metal cations on hydrous manganese oxide using the diffuse double layer model. Appl. Geochem. 2004, 19, 29–53. [Google Scholar] [CrossRef]
- Pretorius, P.J.; Linder, P.W. The adsorption characteristics of δ-manganese dioxide: A collection of diffuse double layer constants for the adsorption of H+, CU2+, NI2+, zn2+, CD2+ and PB2+. Appl. Geochem. 2001, 16, 1067–1082. [Google Scholar] [CrossRef]
- Peacock, C.L.; Sherman, D.M. Sorption of Ni by birnessite: Equilibrium controls on Ni in seawater. Chem. Geol. 2007, 238, 94–106. [Google Scholar] [CrossRef]
- Essington, M.E.; Vergeer, K.A. Adsorption of Antimonate, Phosphate, and Sulfate by Manganese Dioxide: Competitive Effects and Surface Complexation Modeling. Soil Sci. Soc. Am. J. 2015, 79, 803–814. [Google Scholar] [CrossRef]
- Pan, Z.; Li, W.; Fortner, J.D.; Giammar, D.E. Measurement and Surface Complexation Modeling of U(VI) Adsorption to Engineered Iron Oxide Nanoparticles. Environ. Sci. Technol. 2017, 51, 9219–9226. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, T.; Van Riemsdijk, W.H. A Surface Structural Approach to Ion Adsorption: The Charge Distribution (CD) Model. J. Colloid Interface Sci. 1996, 179, 488–508. [Google Scholar] [CrossRef]
- Mangold, J.E.; Park, C.M.; Liljestrand, H.M.; Katz, L.E. Surface complexation modeling of Hg(II) adsorption at the goethite/ water interface using the Charge Distribution Multi-Site Complexation (CD-MUSIC) model. J. Colloid Interface Sci. 2014, 418, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.U.; Frind, E.O.; Blowes, D.W. Multicomponent reactive transport modeling in variably saturated porous media using a generalized formulation for kinetically controlled reactions. Water Resour. Res. 2002, 38, 13-1–13-21. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| k × 103 (s−1), (r2) | ||||
|---|---|---|---|---|
| LC-ICP-MS | Q-XAS | |||
| 60 s Fit | Best Fit | 60 s Fit | Best Fit | |
| From start to cutff time | ||||
| One-parameter | 4.09 (0.81) | 5.64 (0.89) a | 3.90 (0.69) | 34.4 (0.68) c |
| Two-parameter | 3.30 (0.88) | 5.08 (0.91) a | 3.12 (0.78) | 6.25 (0.86) d |
| 60 s window | ||||
| Two-parameter | -- | 0.87 (0.99) b | -- | 6.25 (0.86) d |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schacht, L.; Ginder-Vogel, M. Arsenite Depletion by Manganese Oxides: A Case Study on the Limitations of Observed First Order Rate Constants. Soil Syst. 2018, 2, 39. https://doi.org/10.3390/soilsystems2030039
Schacht L, Ginder-Vogel M. Arsenite Depletion by Manganese Oxides: A Case Study on the Limitations of Observed First Order Rate Constants. Soil Systems. 2018; 2(3):39. https://doi.org/10.3390/soilsystems2030039
Chicago/Turabian StyleSchacht, Lily, and Matthew Ginder-Vogel. 2018. "Arsenite Depletion by Manganese Oxides: A Case Study on the Limitations of Observed First Order Rate Constants" Soil Systems 2, no. 3: 39. https://doi.org/10.3390/soilsystems2030039
APA StyleSchacht, L., & Ginder-Vogel, M. (2018). Arsenite Depletion by Manganese Oxides: A Case Study on the Limitations of Observed First Order Rate Constants. Soil Systems, 2(3), 39. https://doi.org/10.3390/soilsystems2030039