
Changes in Soil Prokaryotic Diversity in Response to Land-Use Changes in Sub-Saharan Africa


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Sampling
2.2. Basic Characteristics of the Soil Samples
2.3. DNA Extraction and Sequencing of 16S rRNA Genes
2.4. Quantitative PCR of 16S rRNA Genes
2.5. Statistical/Prokaryotic Data Analysis
3. Results
3.1. Chemical Properties of the Soils
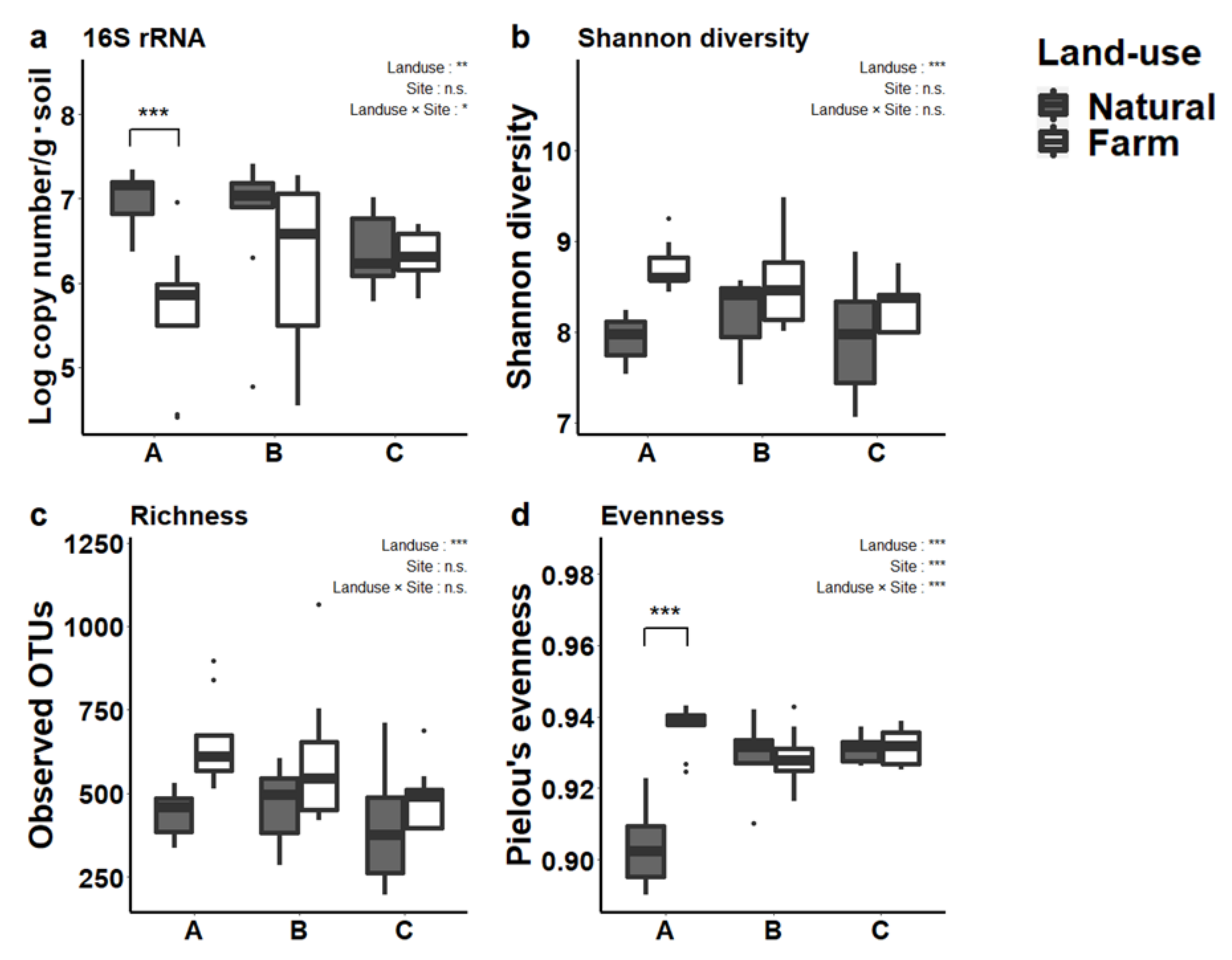
3.2. Prokaryotic Abundance and Diversity
3.3. Identification of Main Prokaryotic Responders towards Management
3.4. Identification of Major Environmental Factors Driving Shifts in Prokaryotic Communities
4. Discussion
4.1. Prokaryotic Community Structures and Diversities Altered by the Cultivation
4.2. Prokaryotic Responders towards Cultivation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tully, K.; Sullivan, C.; Weil, R.; Sanchez, P. The State of Soil Degradation in Sub-Saharan Africa: Baselines, Trajectories, and Solutions. Sustainability 2015, 7, 6523–6552. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.; Abegaz, A.; Matthews, R.B.; Subedi, M.; Orskov, E.R.; Tumwesige, V.; Smith, P. What Is the Potential for Biogas Digesters to Improve Soil Carbon Sequestration in Sub-Saharan Africa? Comparison with Other Uses of Organic Residues. Biomass Bioenergy 2014, 70, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome Definition Re-Visited: Old Concepts and New Challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Allan, E.; Manning, P.; Alt, F.; Binkenstein, J.; Blaser, S.; Blüthgen, N.; Böhm, S.; Grassein, F.; Hölzel, N.; Klaus, V.H.; et al. Land Use Intensification Alters Ecosystem Multifunctionality via Loss of Biodiversity and Changes to Functional Composition. Ecol. Lett. 2015, 18, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Baquerizo, M.; Maestre, F.T.; Reich, P.B.; Jeffries, T.C.; Gaitan, J.J.; Encinar, D.; Berdugo, M.; Campbell, C.D.; Singh, B.K. Microbial Diversity Drives Multifunctionality in Terrestrial Ecosystems. Nat. Commun. 2016, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Schloter, M.; Nannipieri, P.; Sørensen, S.J.; van Elsas, J.D. Microbial Indicators for Soil Quality. Biol. Fertil. Soils 2018, 54, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Baquerizo, M.; Maestre, F.T.; Reich, P.B.; Trivedi, P.; Osanai, Y.; Liu, Y.-R.; Hamonts, K.; Jeffries, T.C.; Singh, B.K. Carbon Content and Climate Variability Drive Global Soil Bacterial Diversity Patterns. Ecol. Monogr. 2016, 86, 373–390. [Google Scholar] [CrossRef]
- Kaiser, K.; Wemheuer, B.; Korolkow, V.; Wemheuer, F.; Nacke, H.; Schöning, I.; Schrumpf, M.; Daniel, R. Driving Forces of Soil Bacterial Community Structure, Diversity, and Function in Temperate Grasslands and Forests. Sci. Rep. 2016, 6, 33696. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wu, J.; Yang, F.; Lei, Y.; Zhang, Q.; Cheng, X. Alterations in Soil Microbial Community Composition and Biomass Following Agricultural Land Use Change. Sci. Rep. 2016, 6, 36587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamgan Nkuekam, G.; Cowan, D.A.; Valverde, A. Arable Agriculture Changes Soil Microbial Communities in the South African Grassland Biome. S. Afr. J. Sci. 2018, 114, 1–5. [Google Scholar] [CrossRef]
- Katulanda, P.M.; Walley, F.L.; Janzen, H.H.; Helgason, B.L. Land Use Legacy Regulates Microbial Community Composition in Transplanted Chernozems. Appl. Soil Ecol. 2018, 129, 13–23. [Google Scholar] [CrossRef]
- Lauber, C.L.; Strickland, M.S.; Bradford, M.A.; Fierer, N. The Influence of Soil Properties on the Structure of Bacterial and Fungal Communities across Land-Use Types. Soil Biol. Biochem. 2008, 40, 2407–2415. [Google Scholar] [CrossRef]
- Hu, S.J.; van Bruggen, A.H.C.; Grünwald, N.J. Dynamics of Bacterial Populations in Relation to Carbon Availability in a Residue-Amended Soil. Appl. Soil Ecol. 1999, 13, 21–30. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Reich, P.B.; Khachane, A.N.; Campbell, C.D.; Thomas, N.; Freitag, T.E.; Abu Al-Soud, W.; Sørensen, S.; Bardgett, R.D.; Singh, B.K. It Is Elemental: Soil Nutrient Stoichiometry Drives Bacterial Diversity: C:N:P Stoichiometry Drives Bacterial Diversity. Environ. Microbiol. 2017, 19, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Dewitte, O.; Jones, A.; Spaargaren, O.; Breuning-Madsen, H.; Brossard, M.; Dampha, A.; Deckers, J.; Gallali, T.; Hallett, S.; Jones, R.; et al. Harmonisation of the Soil Map of Africa at the Continental Scale. Geoderma 2013, 211–212, 138–153. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global Patterns of 16S RRNA Diversity at a Depth of Millions of Sequences per Sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, P. VEGAN, a Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Wang, Y.; Naumann, U.; Wright, S.T.; Warton, D.I. Mvabund—An R Package for Model-Based Analysis of Multivariate Abundance Data. Methods Ecol. Evol. 2012, 3, 471–474. [Google Scholar] [CrossRef]
- Jacquiod, S.; Stenbæk, J.; Santos, S.S.; Winding, A.; Sørensen, S.J.; Priemé, A. Metagenomes Provide Valuable Comparative Information on Soil Microeukaryotes. Res. Microbiol. 2016, 167, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Kuramae, E.E.; Yergeau, E.; Wong, L.C.; Pijl, A.S.; Veen, J.A.; Kowalchuk, G.A. Soil Characteristics More Strongly Influence Soil Bacterial Communities than Land-Use Type. FEMS Microbiol. Ecol. 2012, 79, 12–24. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, D.; Bai, E. Decreasing Soil Microbial Diversity Is Associated with Decreasing Microbial Biomass under Nitrogen Addition. Soil Biol. Biochem. 2018, 120, 126–133. [Google Scholar] [CrossRef]
- Tardy, V.; Spor, A.; Mathieu, O.; Lévèque, J.; Terrat, S.; Plassart, P.; Regnier, T.; Bardgett, R.D.; van der Putten, W.H.; Roggero, P.P.; et al. Shifts in Microbial Diversity through Land Use Intensity as Drivers of Carbon Mineralization in Soil. Soil Biol. Biochem. 2015, 90, 204–213. [Google Scholar] [CrossRef]
- Giller, K.E.; Witter, E.; Mcgrath, S.P. Toxicity of Heavy Metals to Microorganisms and Microbial Processes in Agricultural Soils: A Review. Soil Biol. Biochem. 1998, 30, 1389–1414. [Google Scholar] [CrossRef]
- Zeng, J.; Liu, X.; Song, L.; Lin, X.; Zhang, H.; Shen, C.; Chu, H. Nitrogen Fertilization Directly Affects Soil Bacterial Diversity and Indirectly Affects Bacterial Community Composition. Soil Biol. Biochem. 2016, 92, 41–49. [Google Scholar] [CrossRef]
- Bardgett, R.D.; McAlister, E. The Measurement of Soil Fungal: Bacterial Biomass Ratios as an Indicator of Ecosystem Self-Regulation in Temperate Meadow Grasslands. Biol. Fertil. Soils 1999, 29, 282–290. [Google Scholar] [CrossRef]
- de Vries, F.T.; Manning, P.; Tallowin, J.R.B.; Mortimer, S.R.; Pilgrim, E.S.; Harrison, K.A.; Hobbs, P.J.; Quirk, H.; Shipley, B.; Cornelissen, J.H.C.; et al. Abiotic Drivers and Plant Traits Explain Landscape-Scale Patterns in Soil Microbial Communities. Ecol. Lett. 2012, 15, 1230–1239. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-Based Assessment of Soil PH as a Predictor of Soil Bacterial Community Structure at the Continental Scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Pester, M.; Rattei, T.; Flechl, S.; Gröngröft, A.; Richter, A.; Overmann, J.; Reinhold-Hurek, B.; Loy, A.; Wagner, M. AmoA-Based Consensus Phylogeny of Ammonia-Oxidizing Archaea and Deep Sequencing of AmoA Genes from Soils of Four Different Geographic Regions. Environ. Microbiol. 2012, 14, 525–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chapman, S.J.; Nicol, G.W.; Yao, H. Nitrification and Nitrifiers in Acidic Soils. Soil Biol. Biochem. 2018, 116, 290–301. [Google Scholar] [CrossRef]
- Ramírez, P.B.; Fuentes-Alburquenque, S.; Díez, B.; Vargas, I.; Bonilla, C.A. Soil Microbial Community Responses to Labile Organic Carbon Fractions in Relation to Soil Type and Land Use along a Climate Gradient. Soil Biol. Biochem. 2020, 141, 107692. [Google Scholar] [CrossRef]
- Itakura, M.; Saeki, K.; Omori, H.; Yokoyama, T.; Kaneko, T.; Tabata, S.; Ohwada, T.; Tajima, S.; Uchiumi, T.; Honnma, K.; et al. Genomic Comparison of Bradyrhizobium Japonicum Strains with Different Symbiotic Nitrogen-Fixing Capabilities and Other Bradyrhizobiaceae Members. ISME J. 2009, 3, 326–339. [Google Scholar] [CrossRef]
- Hug, L.A.; Castelle, C.J.; Wrighton, K.C.; Thomas, B.C.; Sharon, I.; Frischkorn, K.R.; Williams, K.H.; Tringe, S.G.; Banfield, J.F. Community Genomic Analyses Constrain the Distribution of Metabolic Traits across the Chloroflexi Phylum and Indicate Roles in Sediment Carbon Cycling. Microbiome 2013, 1, 22. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A Global Atlas of the Dominant Bacteria Found in Soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Norton, J.; Ouyang, Y. Controls and Adaptive Management of Nitrification in Agricultural Soils. Front. Microbiol. 2019, 10, 1931. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohigashi, T.; Schloter, M.; Schulz, S.; Munthali, K.; Uchida, Y. Changes in Soil Prokaryotic Diversity in Response to Land-Use Changes in Sub-Saharan Africa. Soil Syst. 2021, 5, 62. https://doi.org/10.3390/soilsystems5040062
Ohigashi T, Schloter M, Schulz S, Munthali K, Uchida Y. Changes in Soil Prokaryotic Diversity in Response to Land-Use Changes in Sub-Saharan Africa. Soil Systems. 2021; 5(4):62. https://doi.org/10.3390/soilsystems5040062
Chicago/Turabian StyleOhigashi, Takamitsu, Michael Schloter, Stefanie Schulz, Kabenuka Munthali, and Yoshitaka Uchida. 2021. "Changes in Soil Prokaryotic Diversity in Response to Land-Use Changes in Sub-Saharan Africa" Soil Systems 5, no. 4: 62. https://doi.org/10.3390/soilsystems5040062
APA StyleOhigashi, T., Schloter, M., Schulz, S., Munthali, K., & Uchida, Y. (2021). Changes in Soil Prokaryotic Diversity in Response to Land-Use Changes in Sub-Saharan Africa. Soil Systems, 5(4), 62. https://doi.org/10.3390/soilsystems5040062