
Surveillance of Colorectal Cancer (CRC) in Cystic Fibrosis (CF) Patients

 ,
,
Abstract
:1. Introduction
2. Cystic Fibrosis and Colon Cancer
3. Epidemiology
4. Pathophysiology
5. Screening
6. Dysbiosis in Cystic Fibrosis (CFTR Deficiency) Patients
7. CF Patients Are at High Risk for Colorectal Cancer (CRC)
8. CFTR Carriers Are also Susceptible to Gastrointestinal (GI) Cancers
9. Role of Fecal Immunochemical Test (FIT) and Other Fecal Tests
10. Role of Computed Tomography Colonography (CTC)
11. Role of Colonoscopy in CF Patients
12. Role of Colonoscopy in CF Transplanted Patients
13. Role of Bowel Preparation
14. Future Considerations
15. Conclusions
Funding
Conflicts of Interest
References
- Ayoub, F.; Li, H.; Blay, C.; Trillo-Alvarez, C.; Lascano, J.; Morelli, G. Multidisciplinary Care for Cystic Fibrosis Liver Disease: Where Does the Adult Hepatologist Fit In? Clin. Liver Dis. 2019, 14, 187–190. [Google Scholar] [CrossRef]
- O’Sullivan, B.P.; Freedman, S.D. Cystic Fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Gelfond, D.; Borowitz, D. Gastrointestinal complications of Cystic Fibrosis. Clin. Gastroenterol. Hepatol. 2013, 11, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Neglia, J.P.; FitzSimmons, S.C.; Maisonneuve, P.; Schöni, M.H.; Schöni-Affolter, F.; Corey, M.; Lowenfels, A.B. The risk of cancer among patients with Cystic Fibrosis. Cystic Fibrosis and Cancer Study Group. N. Engl. J. Med. 1995, 332, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the Cystic Fibrosis Gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D. Novel personalized therapies for Cystic Fibrosis: Treating the Basic Defect in All Patients. J. Intern. Med. 2015, 277, 155–166. [Google Scholar] [CrossRef]
- Sosnay, P.R.; Raraigh, K.S.; Gibson, R.L. Molecular genetics of cystic fibrosis transmembrane conductance regulator genotype and phenotype. Ped. Clin. N. Am. 2016, 63, 585–598. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Cystic Fibrosis Foundation Patient Registry. Annual Data Report. Bethesda, Maryland. 2017. Available online: https://www.oforg/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf (accessed on 17 March 2019).
- Cystic Fibrosis Foundation. 2017 Patient Registry Annual Data Report. Bethesda, M.D. Cystic Fibrosis Foundation. 2018. Available online: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf (accessed on 17 March 2019).
- Smyth, A.R.; Bell, S.C.; Bojcin, S.; Bryon, M.; Duff, A.; Flume, P.; Kashirskaya, N.; Munck, A.; Ratjen, F.; Schwarzenberg, S.J.; et al. European Cystic Fibrosis Society Standards of Care. Best Practice Guidelines. J. Cyst. Fibrosis. 2014, 13 (Suppl. 1), S23–S42. [Google Scholar] [CrossRef] [Green Version]
- Cystic Fibrosis Foundation. Developing Innovative Gastroenterology Speciality Training (DIGEST) Program. Available online: https://www.cff.org/care/Clinician-Resources/Clinocoan-Awards/Clinician-Career-Development-Aweards/Developing-Innovatove-Gastroenterology-Speciali-Training-DIGEST-Program/ (accessed on 17 March 2019).
- Yamada, A.; Komaki, Y.; Komaki, F.; Micic, D.; Zullow, S.; Sakuraba, A. Risk of gastrointestinal cancers in patients with Cystic Fibrosis: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 758–767. [Google Scholar] [CrossRef]
- Maissonneuve, P.; Marshall, B.C.; Knapp, E.A.; Lowenfels, A.B. Cancer risk in Cystic Fibrosis: A 20-year nationwide study from the United States. J. Nat. Cancer Inst. 2013, 105, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, A.K.; Yanik, E.L.; Marshall, B.C.; Wilschanski, M.; Lynch, C.F.; Austin, A.A.; Copeland, G.; Safaeian, M.; Engels, E.A. Cancer risk among lung transplant recipients with Cystic Fibrosis. J. Cist. Fibros. 2016, 16, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billings, J.L.; Dunitz, J.M.; McAllister, S.; Herzog, T.; Bobr, A.; Khoruts, A. Early Colon screening of adult patients with Cystic Fibrosis reveals high incidence of adenomatous colon polyps. J. Clin. Gastroenterol. 2014, 48, e85–e88. [Google Scholar] [CrossRef] [PubMed]
- Gory, I.; Brown, G.; Wilson, J.; Kemp, W.; Paul, E.; Roberts, S.K. Increased risk of colorectal neoplasia in adult patients with Cystic Fibrosis: A matched case-control study. Scand. J. Gastroenterol. 2014, 49, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.L.; Urbanski, S.J.; Hilsden, R.; Rabin, H.; MacNaughton, W.K.; Beck, P.L. The risk of gastrointestinal malignancies in Cystic Fibrosis: Case report of a patient with a near obstructing villous adenoma found on colon cancer screening and Barrett’s Esophagus. J. Cist. Fibros. 2008, 7, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maissonneuve, P.; FitzSimmons, S.C.; Neglia, J.P.; Campbell, P.W., 3 rd; Lowenfels, A.B. Cancer risk in non transplanted and transplanted Cystic Fibrosis patients: A 10 year study. J. Nat. Cancer Inst. 2003, 95, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.C.; Francois, M.L.; Thomas, H.K.; Radford, K.L.; Hawes, D.S.; Mack, T.L.; Cornwell, R.D.; Maloney, J.D.; De Oliveira, N.C. Colon Cancer in Lung Transplant Recipients with CF: Increased risk and results of screening. J. Cyst. Fibros. 2011, 10, 366–369. [Google Scholar] [CrossRef] [Green Version]
- Hadjiliadis, D.; Khoruts, A.; Zauber, A.G.; Hempstead, S.E.; Maisonneuve, P.; Lowenfels, A.B. Cystic Fibrosis Colorectal Cancer Screening Task Force. Cystic Fibrosis Colorectal Cancer Screening Consensus Recommendations. Gastroenterology 2018, 154, 736–745. [Google Scholar] [CrossRef] [Green Version]
- Than, B.L.N.; Linnekamp, J.F.; Starr, T.K.; Largaespada, D.A.; Rod, A.; Zhang, Y.; Bruner, V.; Abrahante, J.; Schumann, A.; Luczak, T.; et al. CFTR is a tumor suppressor gene in murine and human intestinal cancer. Oncogene 2016, 35, 4179–4187. [Google Scholar] [CrossRef]
- Gustafsson, J.K.; Ermund, A.; Ambort, D.; Johansson, M.E.; Nilsson, H.E.; Thorell, K.; Hebert, H.; Sjövall, H.; Hansson, G.C. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med. 2012, 209, 1263–1272. [Google Scholar] [CrossRef] [Green Version]
- Norkina, O.; Burnett, T.G.; De Lisle, R.C. Bacterial overgrowth in the cystic fibrosis transmembrane conductance regulator null mouse small intestine. Infect. Immun. 2004, 72, 6040–6049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushi, L.H.; Doyle, C.; McCullough, M.; Rock, C.L.; Demark-Wahnefried, W.; Bandera, E.V.; Gapstur, S.; Patel, A.V.; Andrews, K.; Gansler, T. American Cancer Society 2010 Nutrition and Physical Activity Guidelines Advisory Committee. American Cancer Society Guidelines on Nutrition and Physical Activity for Cancer prevention: Reducing the risk of cancer with healthy food choices and physical activity. CA Cancer J. Clin. 2012, 62, 30–67. [Google Scholar] [PubMed] [Green Version]
- Mima, K.; Ogino, S.; Nakagawa, S.; Sawayama, H.; Kinoshita, K.; Krashima, R.; Ishimoto, T.; Imai, K.; Iwatsuki, M.; Hashimoto, D.; et al. The role of intestinal bacteria in the development and progression of gastrointestinal neoplasms. Surg. Oncol. 2017, 26, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.S.; Ernst, S.; Tang, X.X.; Karp, P.H.; Parker, C.P.; Ostedgaard, L.S.; Welsh, J.M. Relationships among CFTR expression, HCO3 secretion and host defense may inform gene and cell based Cystioc Fibrosis therapy. Proc. Nat. Acad. Sci. USA 2016, 113, 5382–5387. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; Welsh, M.J.; Zabner, J.; Polgreen, P.M. Cystic Fibrosis carriers are at increased risk for a wide range of Cystic Fibrosis related conditions. Proc. Nat. Acad. Sci. USA 2020, 117, 1621–1627. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, S.M.; Chmiel, J.F.; Sternen, D.L.; Cheng, E.; Gibson, R.L.; Marshall, S.G.; Cutting, G.R. Clinical practice and genetic counseling for Cystic Fibrosis and CFTR- related disorders. Genet. Med. 2008, 10, 851–868. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J. Identification of the Cystic Fibrosis gene: Cloning and characterization of complementary cDNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Scott, P.; Anderson, K.; Singhania, M.; Cormier, R. Cystic Fibrosis, CFTR and Colorectal Cancer. Intern. J. Mol. Sci. 2020, 21, 2891. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef]
- Jakab, R.L.; Collaco, A.M.; Ameen, N.A. Physiological relevance of cell-specific distribution patterns of CFTR, NKCC1, NBCe1 and NHE3 along the crypt-villus axis in the intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G82–G98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van der Born, M.; Cozijnesen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Sorscher, E.J. Cystic Fibrosis in “Harrison’s Principles of Internal Medicine; Kasper, D., Fauci, A., Hauser, S., Longo, D., Jameson, J.L., Loscalzo, J., Eds.; McGraw-Hill Education: New York, NY, USA, 2015. [Google Scholar]
- Liou, T.G. The clinical biology of Cystic Fibrosis TRansmembrane Regulator Protein: Its role and function in extrapulmonary disease. Chest 2019, 155, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Guggino, W.B.; Styanton, B.A. New insights into Cysatic Fibrosis: Molecular switches that regulate CFTR. Nat. Rev. Med. Cell Biol. 2006, 7, 426–436. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; Van Es, J.H.; van de Wetering, M.; Begthel, H.; van der Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevres, H. Crypt stme cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Niccum, D.E.; Billigs, J.L.; Dunitz, J.M.; Khoruts, A. Colonoscopic screening shows increased early incidence and progression of adenomas in Cystic Fibrosis. J. Cist. Fibros. 2016, 15, 548–553. [Google Scholar] [CrossRef] [Green Version]
- Levin, B.; Lieberman, D.A.; McFarland, B.; Smith, R.A.; Brooks, D.; Andrews, K.S.; Dash, C.; Giardiello, F.M.; Glick, S.; Levin, T.R.; et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: A joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J. Clin. 2008, 58, 130–160. [Google Scholar] [CrossRef] [Green Version]
- Lindor, N.M.; Petersen, G.M.; Hadley, D.W.; Kinney, A.Y.; Miesfeldt, S.; Lu, K.H.; Lynch, P.; Burke, W.; Press, N. Reccomendations for the care of individuals with an inherited predisposition to Lynch Syndrome: A systematic review. JAMA 2006, 296, 1507–1517. [Google Scholar] [CrossRef]
- Bonadona, V.; Bonaiti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutationsin MLH1, MSH2 and MSH6 genes in Lynch Syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation Patient Registry 2015Annual Data Report, Bethesda, Maryland. Available online: https//www.cff.org/our-research/CF-patient-registry/2015-patient-registry-annual-data-report.pdf 2016 (accessed on 17 March 2019).
- De Lisle, R.C.; Borowitz, D. The Cystic Fibrosis Intestine. Cold Spring Harb. Prespeciv. Med. 2013, 3, a009753. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.V.; Goldfarb, K.C.; Wild, Y.K.; Kong, W.; De Lisle, R.C.; Brodie, E.L. Cystic Fibrosis Transmembrane conductance regulator knockout mice exhibit aberrant gastrointestinal microbiota. Gut Microbes 2013, 4, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeker, S.M.; Mears, K.S.; Sangwan, N.; Brittnacher, M.J.; Weiss, E.J.; Treuting, P.M.; Tolley, N.; Pope, C.E.; Hager, K.R.; Vo, A.T.; et al. CFTR dysregulation drives active selection of the gut microbiome. PLoS Pathog. 2002, 16, e1008251. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The intestinal microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A functional crosstalk. Front. Physiol. 2018, 9, 1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cystic Fibrosi Foundation. Patient Registry. 2019. Available online: https//www.cff.org/ (accessed on 1 December 2019).
- Cystic Fibrosis Foundation. Carrier testing for Cystic Fibrosis 2019. Available online: https://www.cff.org/ (accessed on 1 December 2019).
- Gini, A.; Zauber, A.G.; Cenin, D.R.; Omidvari, A.H.; Hempstead, S.E.; Fink, A.K.; Lowenfels, A.B.; Lansdorp-Vogelaar, I. Cost effectiveness of screening individuals with Cystic Fibrosis for Colorectal Cancer. Gastroenterology 2018, 154, 556–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, J.M.; Mahan, K.; Mettler, T.; Dunitz, J.M.; Khoruts, A. Case report of synchronous post –lung transplant colon cancers in the era of colorectal cancer screening recommendation in Cystic Fibrosis: Screening “too early” before it’s too late. BMC Gastroenterol. 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkin, W.S.; Valori, R.; Kuipers, E.J.; Hoff, G.; Senore, C.; Segnan, N.; Jover, R.; Schmiegel, W.; Lambert, R.; Pox, C. European guidelines for quality assurance in colorectal cancer screening and diagnosis. First Edition-Colonoscopic surveillance following adenoma removal. Endoscopy 2012, 44 (Suppl. 3), SE151–SE163. [Google Scholar] [CrossRef] [Green Version]
- Rex, D.K. Optimal Bowel preparation-a practical guide for clinicians. Nast. Rev. Gastroenterol. Hepatol. 2014, 11, 419–425. [Google Scholar] [CrossRef]
- Hassan, C.; East, J.; Radaelli, F.; Spada, C.; Benamouzig, R.; Bisschops, R.; Bretthauer, M.; Dekker, E.; Dinis-Ribeiro, M.; Ferlitsch, M.; et al. Bowel preparation for Colonoscopy: European Society of Gastrointestinal Endoscopy (ESGE) guideline–Update 2019. Endoscopy 2019, 51, 775–794. [Google Scholar] [CrossRef] [Green Version]
- Samarasena, J.B.; Muthusamy, V.R.; Jamal, M.M. Split-Dosed MiraLAX/Gatorade Is an Effective, Safe, and Tolerable Option for Bowel Preparation in Low-Risk Patients: A Randomized Controlled Study. Am. J. Gastroenterol. 2012, 107, 1036–1042. [Google Scholar] [CrossRef]
- Nagler, J.; Poppers, D.; Turetz, M. Severe Hyponatremia and Seizure Following a Polyethylene Glycol-Based Bowel Preparation for Colonoscopy. J. Clin. Gastroenterol. 2006, 40, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Kurnar, S.; Thosani, N.; Ladabaum, U.; Friedland, S.; Chen, A.M.; Kochar, R.; Banerjee, S. Adenoma Miss Rates Associated With a 3-minute Versus 6-minute Colonoscopy Withdrawal Time: A Prospective, Randomized Trial. Gastrointest Endosc. 2017, 85, 1273–1280. [Google Scholar]
- ASGE Standards of Practice Committee; Fisher, D.A.; Maple, J.T.; Ben-Menachem, T.; Cash, B.D.; Decker, G.A.; Early, D.S.; Evans, J.A.; Fanelli, R.D.; Fukami, N.; et al. Complications of Colonoscopy. Gastrointest. Endosc. 2011, 74, 745–752. [Google Scholar]
- Matson, A.G.; Bunting, J.P.; Kaul, A.; Smith, D.J.; Stonestreet, J.; Herd, K.; Hodgson, R.S.; Bell, S.C. A non-randomised single center cohort study, comparing standard and modified bowel preparations, in adults with Cystic Fibrosis requiring Colonscopy. BMC Gastroenterol. 2019, 19, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queensland Health. Queensland Health. Queensland Bowel Cancer Screening Program Bowel Preparation Descriptors. In Queensland Bowel Cancer Screening Program State Coordination Unit; Queenland, P.I., Ed.; Queenland Health: Brisbane, Australia, 2012. [Google Scholar]
- Lai, E.J.; Calderwood, A.H.; Doros, G.; Fix, O.K.; Jacobson, B.C. The Boston Bowel preparation scale: A valid and reliable instrument for colonoscopy-oriented research. Gastrointest. Endosc. 2009, 69 Pt 2, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Fajac, I.; Wainwright, C.E. New treatments targeting the basic defects in Cystic Fibrosis. Presse Med. 2017, 46 Pt 2, e165–e175. [Google Scholar] [CrossRef]
- Hou, Y.; Guan, X.; Yang, Z.; Li, C. Emerging role of Cystic Fibrosis Transmembrane Conductance Regulator- an epithelial Chloride channel in Gastrointestinal Cancers. World J. GastroInt. Oncol. 2016, 8, 282–288. [Google Scholar] [CrossRef]
- Ooi, C.Y.; Syed, S.A.; Rossi, L.; Garg, M.; Needham, B.; Avolio, J.; Young, K.; Surette, M.G.; Gonska, T. Impact of CFTR modulation with Ivacaftor on Gut Microbiota and Intestinal Inflammation. Sci. Rep. 2018, 8, 17834. [Google Scholar] [CrossRef] [Green Version]
- Werlin, S.L.; Benuri-Silbiger, I.; Kerem, E.; Adler, S.N.; Goldin, E.; Zimmerman, J.; Malka, N.; Cohen, L.; Armoni, S.; Yatzkan-Israelit, Y.; et al. Evidence of Intestinal inflammation in patients with Cystic Fibrosis. J. Ped. Gastroenterol. Nutr. 2010, 51, 304–308. [Google Scholar] [CrossRef]
- Lochhead, P.; Chan, A.T. Statins and Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2013, 11, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.C.S.; Huang, J.; Lok, V.; Wang, J.; Fung, F.; Ding, H.; Zheng, Z.J. Differences in Incidence and Mortality Trends of Colorectal Cancer, Worldwide, Based on Sex, Age, and Anatomic Location. Clin. Gastroenterol. Hepatol. 2021, 19, 955–966.e61. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| (1) Colonoscopy should be considered the most important screening examination for CRC screening in CF patients |
| (2) Computed Tomography Colonography (CTC), Blood Occult and DNA stool screening test, sigmoidoscopy are not recommended as screening tests for CF patients |
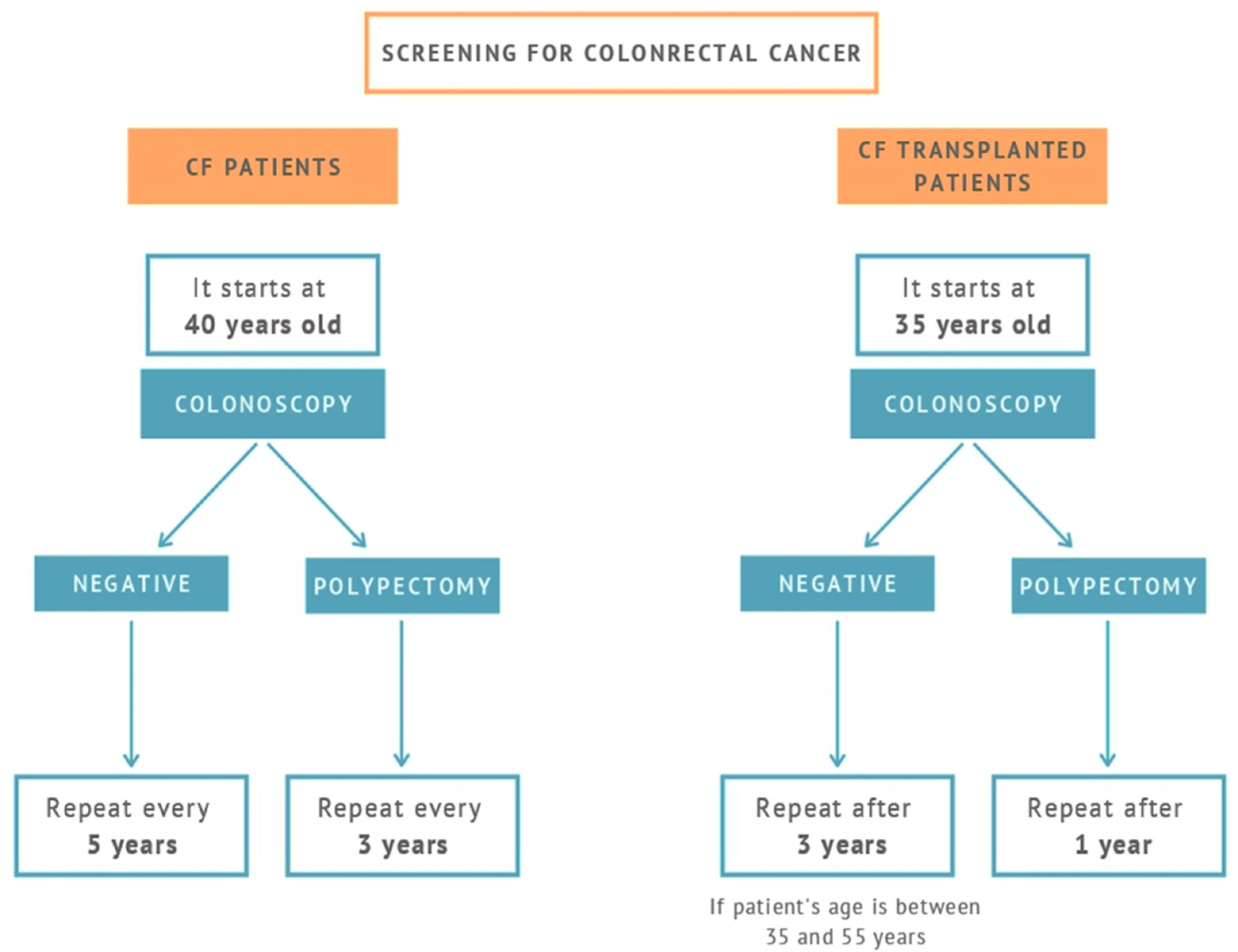
| (3) CRC screening should be started at age 40 in CF patients and rescreening should continue every 5 years |
| (4) In presence of adenomatous polyps, a colonoscopy should be performed every 3 years and annually in presence of large and multiple polyps |
| (5) In patients with CF of 30 years of age and subjected to solid organ transplantation, colonoscopy is indicated after 2 years from transplantation except in subjects with negative colonoscopy performed in the last 5 years |
| (6) In case of diagnosis of adenomatous polyps in transplanted patients, surveillance colonoscopy should be performed every 3 years and shorter intervals if adenomatous polyps are large (>1 cm in diameter) and multiple (if it is possible, every year, and if negative colonoscopy, every 3 years) |
| (7) CF foundation recommends an intensive regimen for bowel preparation in CF patients with 3–4 washes (minimum of 1 l purgative per wash) with the last wash occurring within 4–6 h before examination |
| (8) CF Foundation recommends that all decisions about CRC screening and CF patient should be managed by CF health care professional and endoscopist |
| (9) CF Foundation recommends that all decisions on CRC screening and surveillance in CF patients be always based on a common decision between provider and individual with CF about treatment, comorbidities, safety and quality of life |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ingravalle, F.; Casella, G.; Ingravalle, A.; Monti, C.; De Salvatore, F.; Stillitano, D.; Villanacci, V. Surveillance of Colorectal Cancer (CRC) in Cystic Fibrosis (CF) Patients. Gastrointest. Disord. 2021, 3, 84-95. https://doi.org/10.3390/gidisord3020009
Ingravalle F, Casella G, Ingravalle A, Monti C, De Salvatore F, Stillitano D, Villanacci V. Surveillance of Colorectal Cancer (CRC) in Cystic Fibrosis (CF) Patients. Gastrointestinal Disorders. 2021; 3(2):84-95. https://doi.org/10.3390/gidisord3020009
Chicago/Turabian StyleIngravalle, Fabio, Giovanni Casella, Adriana Ingravalle, Claudio Monti, Federica De Salvatore, Domenico Stillitano, and Vincenzo Villanacci. 2021. "Surveillance of Colorectal Cancer (CRC) in Cystic Fibrosis (CF) Patients" Gastrointestinal Disorders 3, no. 2: 84-95. https://doi.org/10.3390/gidisord3020009
APA StyleIngravalle, F., Casella, G., Ingravalle, A., Monti, C., De Salvatore, F., Stillitano, D., & Villanacci, V. (2021). Surveillance of Colorectal Cancer (CRC) in Cystic Fibrosis (CF) Patients. Gastrointestinal Disorders, 3(2), 84-95. https://doi.org/10.3390/gidisord3020009