
Tribbles Gene Expression Profiles in Colorectal Cancer

 ,
, 
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Tribbles Expression in Colon Cancer Tissues and Cell Lines
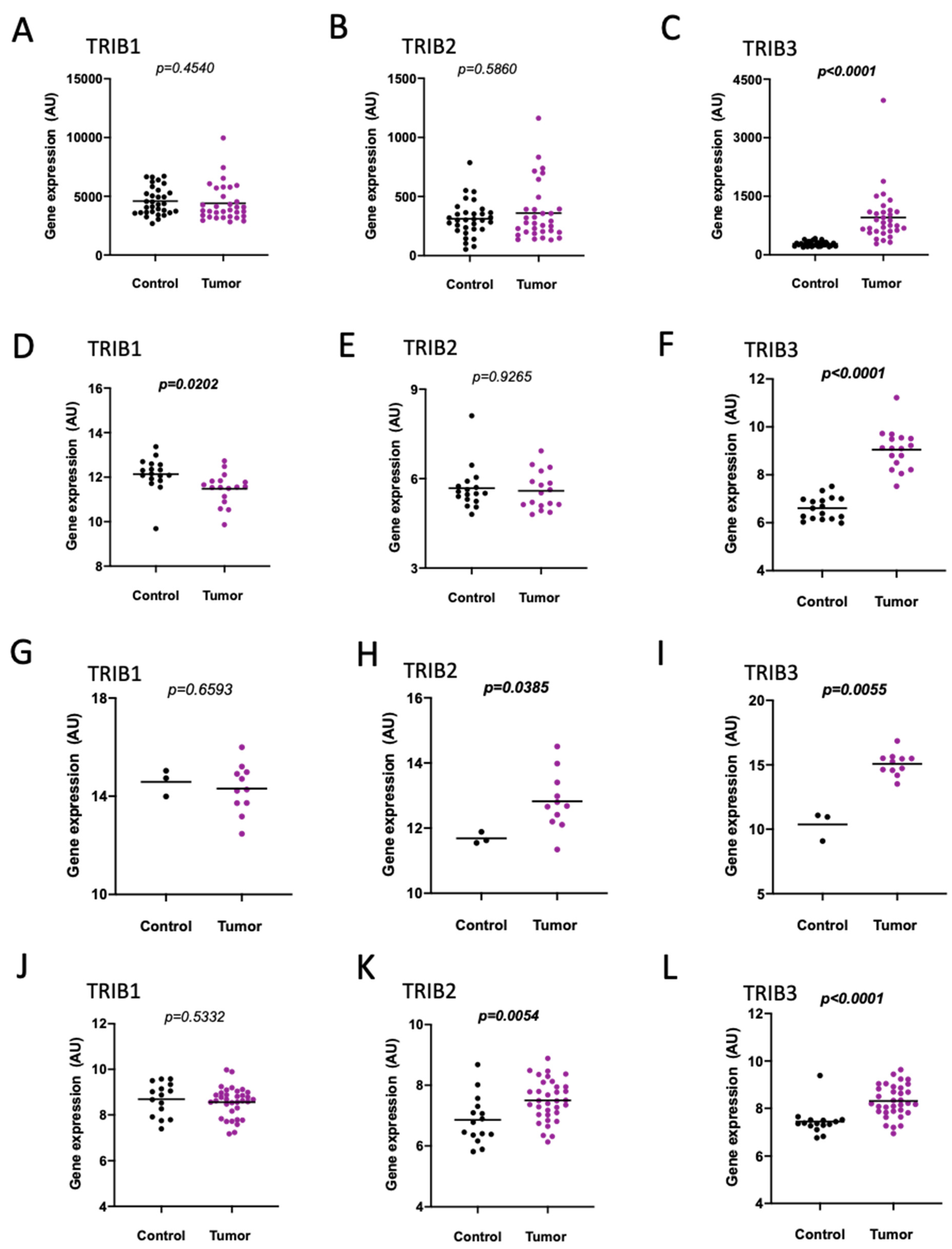
2.1.1. Colon and Rectal Cancer Tissues Compared to Controls
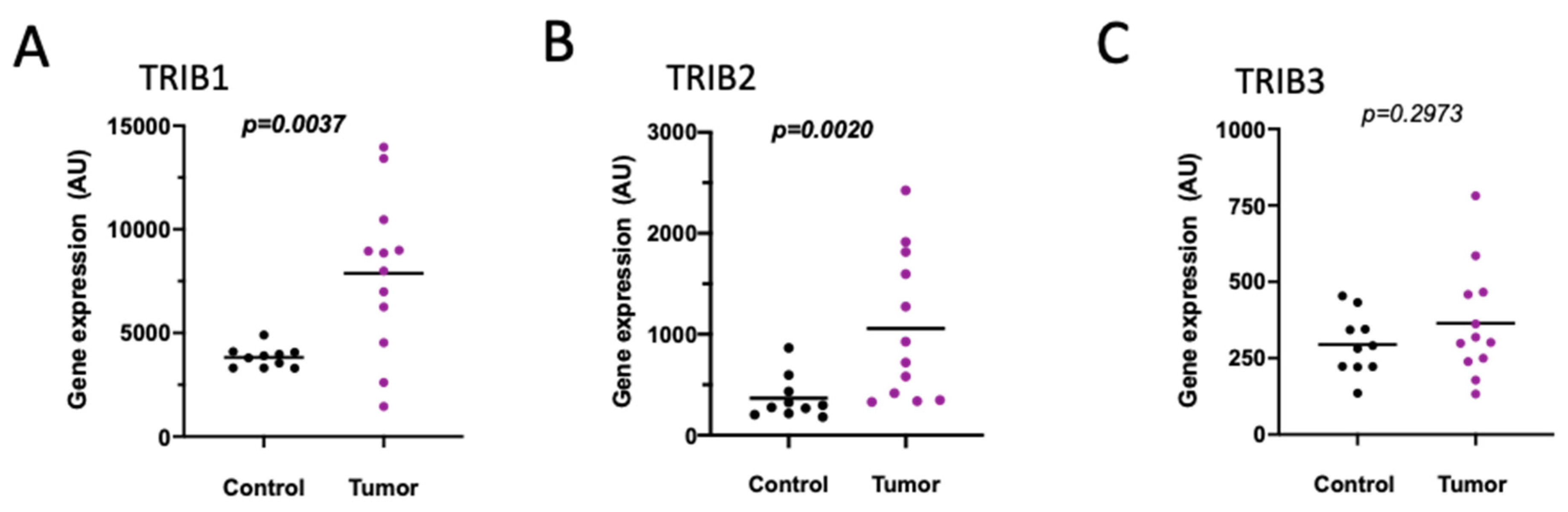
2.1.2. Colon Cancer Cells Lines
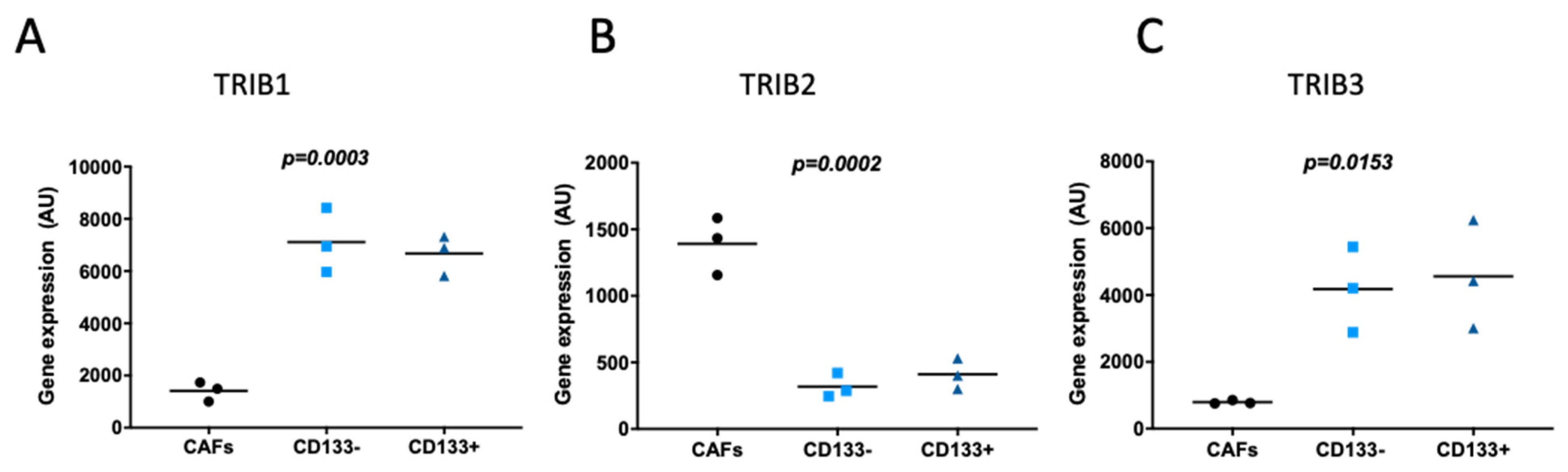
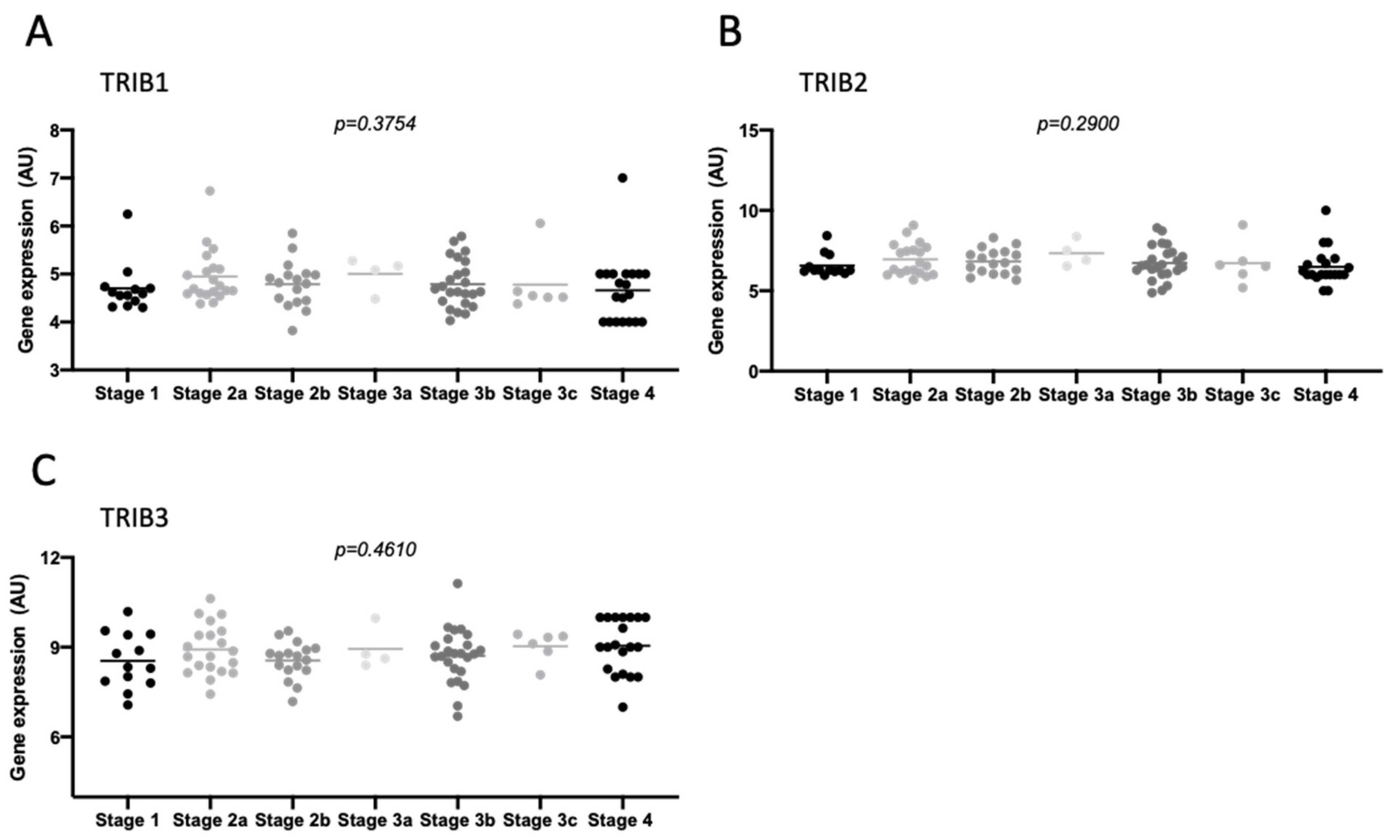
2.2. Tribbles Expression Association to Colon Cancer Progression, Staging, and Metastasis
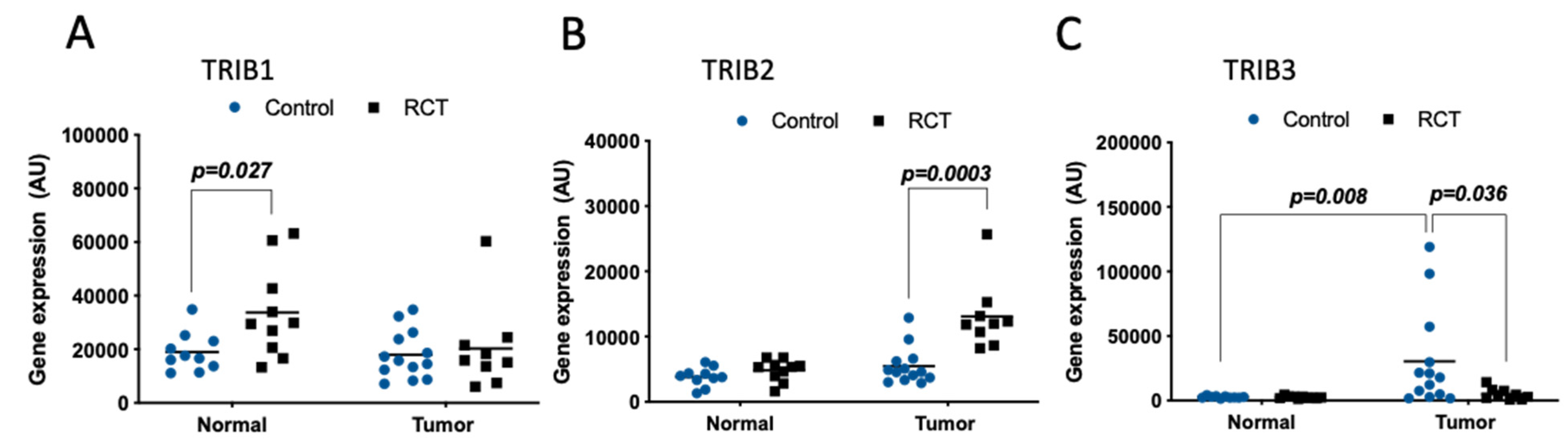
2.3. Tribbles Expression and Its Association to Colon Cancer Relapse
2.4. Tribbles Expression Association to Colon Cancer Drug-Resistance
2.5. Tribbles Transcriptional Regulation in Colon Cancer
2.5.1. Tribbles Regulation in Response to Protein Modulation
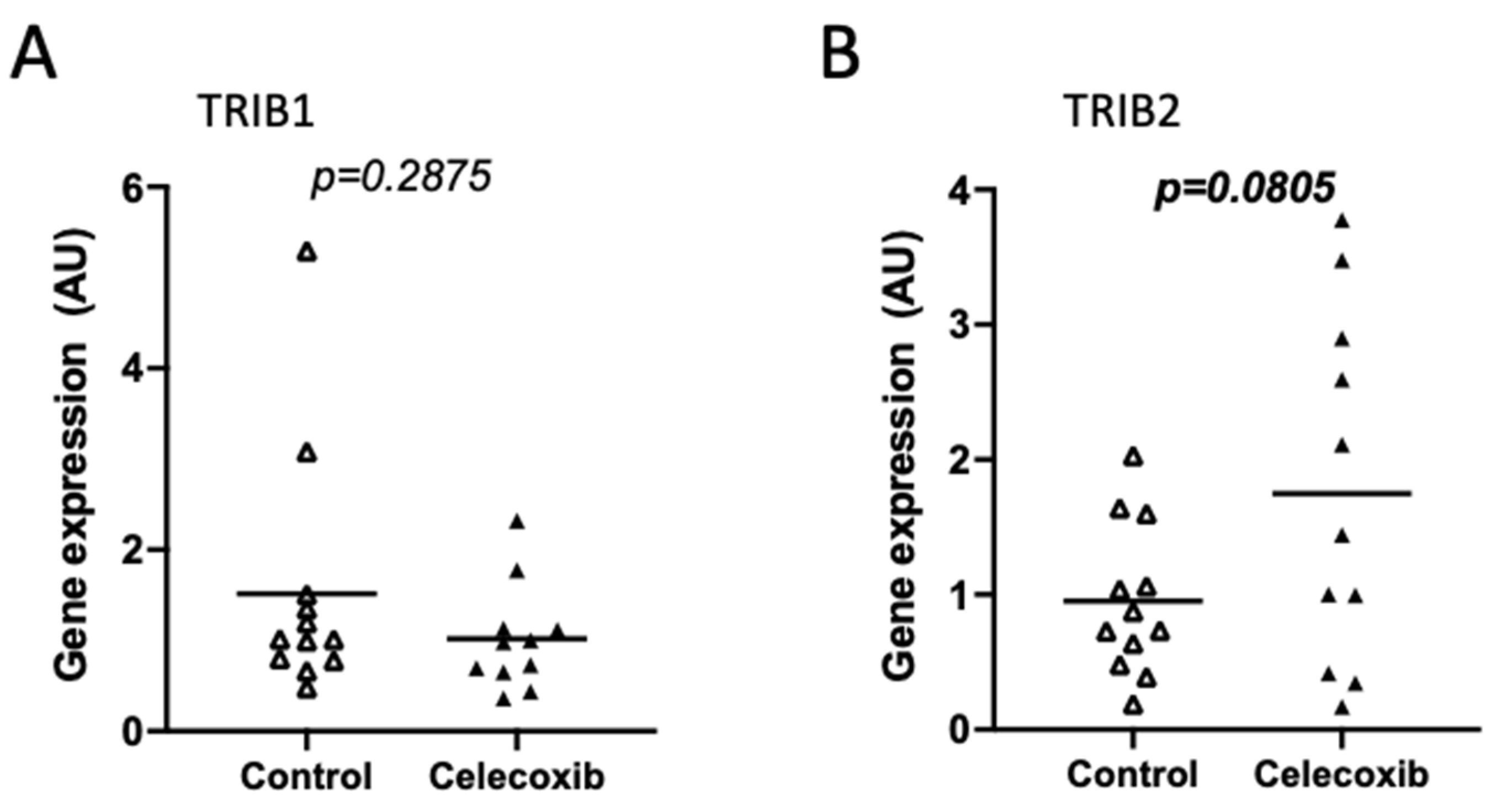
2.5.2. Tribbles Regulation in Response to Pharmacological Treatments
3. Discussion
4. Materials and Methods
4.1. Sources of Data
4.2. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Arends, J.W. Molecular interactions in the Vogelstein model of colorectal carcinoma. J. Pathol. 2000, 190, 412–416. [Google Scholar] [CrossRef]
- Tariq, K.; Ghias, K. Colorectal cancer carcinogenesis: A review of mechanisms. Cancer Biol. Med. 2016, 13, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. (Eds.) World Cancer Report: Cancer Research for Cancer Prevention; IARC Press: Lyon, France, 2020; ISBN 9789283204299. [Google Scholar]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Kim, T. Il Serrated neoplasia pathway as an alternative route of colorectal cancer carcinogenesis. Intest. Res. 2018, 16, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Kambara, T.; Simms, L.A.; Whitehall, V.L.J.; Spring, K.J.; Wynter, C.V.A.; Walsh, M.D.; Barker, M.A.; Arnold, S.; McGivern, A.; Matsubara, N.; et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 2004, 53, 1137–1144. [Google Scholar] [CrossRef] [Green Version]
- Iino, H.; Jass, J.R.; Simms, L.A.; Young, J.; Leggett, B.; Ajioka, Y.; Watanabe, H. DNA microsatellite instability in hyperplastic polyps, serrated adenomas, and mixed polyps: A mild mutator pathway for colorectal cancer? J. Clin. Pathol. 1999, 52, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, N.J.; Ward, R.L. Sporadic colorectal cancers with microsatellite instability and their possible origin in hyperplastic polyps and serrated adenomas. J. Natl. Cancer Inst. 2001, 93, 1307–1313. [Google Scholar] [CrossRef]
- Mäkinen, M.J.; George, S.M.; Jernvall, P.; Mäkelä, J.; Vihko, P.; Karttunen, T.J. Colorectal carcinoma associated with serrated adenoma—Prevalence, histological features, and prognosis. J. Pathol. 2001, 193, 286–294. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Sawyer, E.J.; Cerar, A.; Hanby, A.M.; Gorman, P.; Arends, M.; Talbot, I.C.; Tomlinson, I.P.M. Molecular characteristics of serrated adenomas of the colorectum. Gut 2002, 51, 200–206. [Google Scholar] [CrossRef]
- Laiho, P.; Kokko, A.; Vanharanta, S.; Salovaara, R.; Sammalkorpi, H.; Järvinen, H.; Mecklin, J.-P.; Karttunen, T.J.; Tuppurainen, K.; Davalos, V.; et al. Serrated carcinomas form a subclass of colorectal cancer with distinct molecular basis. Oncogene 2007, 26, 312–320. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society Cancer Facts & Figures 2021. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html (accessed on 29 October 2021).
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2016, 27, 284–298. [Google Scholar] [CrossRef] [Green Version]
- Richmond, L.; Keeshan, K. Pseudokinases: A tribble-edged sword. FEBS J. 2020, 287, 4170–4182. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, T.; Nakamura, T. Tribbles in disease: Signaling pathways important for cellular function and neoplastic transformation. Cancer Sci. 2011, 102, 1115–1122. [Google Scholar] [CrossRef]
- Mayoral-Varo, V.; Jiménez, L.; Link, W. The Critical Role of TRIB2 in Cancer and Therapy Resistance. Cancers 2021, 13, 2701. [Google Scholar] [CrossRef]
- Stefanovska, B.; André, F.; Fromigué, O. Tribbles Pseudokinase 3 Regulation and Contribution to Cancer. Cancers 2021, 13, 1822. [Google Scholar] [CrossRef]
- Ferreira, B.I.; Santos, B.; Link, W.; De Sousa-Coelho, A.L. Tribbles Pseudokinases in Colorectal Cancer. Cancers 2021, 13, 2825. [Google Scholar] [CrossRef]
- Hou, Z.; Guo, K.; Sun, X.; Hu, F.; Chen, Q.; Luo, X.; Wang, G.; Hu, J.; Sun, L. TRIB2 functions as novel oncogene in colorectal cancer by blocking cellular senescence through AP4/p21 signaling. Mol. Cancer 2018, 17, 172. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, N.; Pang, B.; Tong, D.; Sun, D.; Sun, H.; Zhang, C.; Sun, W.; Meng, X.; Bai, J.; et al. TRIB1 promotes colorectal cancer cell migration and invasion through activation MMP-2 via FAK/Src and ERK pathways. Oncotarget 2017, 8, 47931–47942. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, W.; Luo, L.; Han, K.; Liu, R.; Wei, S.; Guo, X. Long noncoding RNA TUG1 regulates the progression of colorectal cancer through miR-542-3p/TRIB2 axis and Wnt/β-catenin pathway. Diagn. Pathol. 2021, 16, 47. [Google Scholar] [CrossRef]
- Dedhia, P.H.; Keeshan, K.; Uljon, S.; Xu, L.; Vega, M.E.; Shestova, O.; Zaks-Zilberman, M.; Romany, C.; Blacklow, S.C.; Pear, W.S. Differential ability of Tribbles family members to promote degradation of C/EBPα and induce acute myelogenous leukemia. Blood 2010, 116, 1321–1328. [Google Scholar] [CrossRef]
- Keeshan, K.; Bailis, W.; Dedhia, P.H.; Vega, M.E.; Shestova, O.; Xu, L.; Toscano, K.; Uljon, S.N.; Blacklow, S.C.; Pear, W.S. Transformation by Tribbles homolog 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood 2010, 116, 4948–4957. [Google Scholar] [CrossRef] [Green Version]
- Salomé, M.; Hopcroft, L.; Keeshan, K. Inverse and correlative relationships between TRIBBLES genes indicate non-redundant functions during normal and malignant hemopoiesis. Exp. Hematol. 2018, 66, 63–78.e13. [Google Scholar] [CrossRef] [Green Version]
- Guan, H.; Shuaib, A.; De Leon, D.D.; Angyal, A.; Salazar, M.; Velasco, G.; Holcombe, M.; Dower, S.K.; Kiss-Toth, E. Competition between members of the tribbles pseudokinase protein family shapes their interactions with mitogen activated protein kinase pathways. Sci. Rep. 2016, 6, 32667. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Sabates-Bellver, J.; Van der Flier, L.G.; de Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 2007, 5, 1263–1275. [Google Scholar] [CrossRef] [Green Version]
- Khamas, A.; Ishikawa, T.; Shimokawa, K.; Mogushi, K.; Iida, S.; Ishiguro, M.; Mizushima, H.; Tanaka, H.; Uetake, H.; Sugihara, K. Screening for epigenetically masked genes in colorectal cancer Using 5-Aza-2′-deoxycytidine, microarray and gene expression profile. Cancer Genom. Proteom. 2012, 9, 67–75. [Google Scholar]
- Ågesen, T.H.; Berg, M.; Clancy, T.; Thiis-Evensen, E.; Cekaite, L.; Lind, G.E.; Nesland, J.M.; Bakka, A.; Mala, T.; Hauss, H.J.; et al. CLC and IFNAR1 are differentially expressed and a global immunity score is distinct between early- and late-onset colorectal cancer. Genes Immun. 2011, 12, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Danielsen, S.A.; Cekaite, L.; Ågesen, T.H.; Sveen, A.; Nesbakken, A.; Thiis-Evensen, E.; Skotheim, R.I.; Lind, G.E.; Lothe, R.A. Phospholipase C isozymes are deregulated in colorectal cancer—Insights gained from gene set enrichment analysis of the transcriptome. PLoS ONE 2011, 6, e24419. [Google Scholar] [CrossRef] [Green Version]
- Alhopuro, P.; Sammalkorpi, H.; Niittymäki, I.; Biström, M.; Raitila, A.; Saharinen, J.; Nousiainen, K.; Lehtonen, H.J.; Heliövaara, E.; Puhakka, J.; et al. Candidate driver genes in microsatellite-unstable colorectal cancer. Int. J. Cancer 2012, 130, 1558–1566. [Google Scholar] [CrossRef]
- Hong, Y.; Ho, K.S.; Eu, K.W.; Cheah, P.Y. A susceptibility gene set for early onset colorectal cancer that integrates diverse signaling pathways: Implication for tumorigenesis. Clin. Cancer Res. 2007, 13, 1107–1114. [Google Scholar] [CrossRef] [Green Version]
- Berg, M.; Danielsen, S.A.; Ahlquist, T.; Merok, M.A.; Ågesen, T.H.; Vatn, M.H.; Mala, T.; Sjo, O.H.; Bakka, A.; Moberg, I.; et al. DNA Sequence Profiles of the Colorectal Cancer Critical Gene Set KRAS-BRAF-PIK3CA-PTEN-TP53 Related to Age at Disease Onset. PLoS ONE 2010, 5, e13978. [Google Scholar] [CrossRef]
- Berg, M.; Ågesen, T.H.; Thiis-Evensen, E.; Merok, M.A.; Teixeira, M.R.; Vatn, M.H.; Nesbakken, A.; Skotheim, R.I.; Lothe, R.A. Distinct high resolution genome profiles of early onset and late onset colorectal cancer integrated with gene expression data identify candidate susceptibility loci. Mol. Cancer 2010, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Snipstad, K.; Fenton, C.G.; Kjaeve, J.; Cui, G.; Anderssen, E.; Paulssen, R.H. New specific molecular targets for radio-chemotherapy of rectal cancer. Mol. Oncol. 2010, 4, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Hervieu, C.; Christou, N.; Battu, S.; Mathonnet, M. The Role of Cancer Stem Cells in Colorectal Cancer: From the Basics to Novel Clinical Trials. Cancers 2021, 13, 1092. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Chao, C.; Carmical, J.R.; Ives, K.L.; Wood, T.G.; Aronson, J.F.; Gomez, G.A.; Djukom, C.D.; Hellmich, M.R. CD133+ colon cancer cells are more interactive with the tumor microenvironment than CD133-cells. Lab. Investig. 2012, 92, 420–436. [Google Scholar] [CrossRef] [Green Version]
- Pfister, T.D.; Reinhold, W.C.; Agama, K.; Gupta, S.; Khin, S.A.; Kinders, R.J.; Parchment, R.E.; Tomaszewski, J.E.; Doroshow, J.H.; Pommier, Y. Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Mol. Cancer Ther. 2009, 8, 1878–1884. [Google Scholar] [CrossRef] [Green Version]
- Kohn, K.W.; Zeeberg, B.M.; Reinhold, W.C.; Pommier, Y. Gene expression correlations in human cancer cell lines define molecular interaction networks for epithelial phenotype. PLoS ONE 2014, 9, e99269. [Google Scholar] [CrossRef] [Green Version]
- Reinhold, W.C.; Sunshine, M.; Varma, S.; Doroshow, J.H.; Pommier, Y. Using CellMiner 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60. Clin. Cancer Res. 2015, 21, 3841–3852. [Google Scholar] [CrossRef] [Green Version]
- Giovinazzi, S.; Sirleto, P.; Aksenova, V.; Morozov, V.M.; Zori, R.; Reinhold, W.C.; Ishov, A.M. Usp7 protects genomic stability by regulating Bub3. Oncotarget 2014, 5, 3728–3742. [Google Scholar] [CrossRef] [Green Version]
- Provenzani, A.; Fronza, R.; Loreni, F.; Pascale, A.; Amadio, M.; Quattrone, A. Global alterations in mRNA polysomal recruitment in a cell model of colorectal cancer progression to metastasis. Carcinogenesis 2006, 27, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, S.; Ishikawa, T.; Iida, S.; Ishiguro, M.; Mogushi, K.; Mizushima, H.; Uetake, H.; Tanaka, H.; Sugihara, K. Clinical significance of osteoprotegerin expression in human colorectal cancer. Clin. Cancer Res. 2011, 17, 2444–2450. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, S.; Midorikawa, Y.; Takahashi, T.; Yagi, K.; Takayama, T.; Yoshida, K.; Sugiyama, Y.; Aburatani, H. Potential responders to FOLFOX therapy for colorectal cancer by Random Forests analysis. Br. J. Cancer 2012, 106, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Menyhart, O.; Kakisaka, T.; Pongor, L.S.; Uetake, H.; Goel, A.; Győrffy, B. Uncovering Potential Therapeutic Targets in Colorectal Cancer by Deciphering Mutational Status and Expression of Druggable Oncogenes. Cancers 2019, 11, 983. [Google Scholar] [CrossRef] [Green Version]
- Bandrés, E.; Malumbres, R.; Cubedo, E.; Honorato, B.; Zarate, R.; Labarga, A.; Gabisu, U.; Sola, J.J.; García-Foncillas, J. A gene signature of 8 genes could identify the risk of recurrence and progression in Dukes’ B colon cancer patients. Oncol. Rep. 2007, 17, 1089–1094. [Google Scholar] [CrossRef] [Green Version]
- Gröne, J.; Lenze, D.; Jurinovic, V.; Hummel, M.; Seidel, H.; Leder, G.; Beckmann, G.; Sommer, A.; Grützmann, R.; Pilarsky, C.; et al. Molecular profiles and clinical outcome of stage UICC II colon cancer patients. Int. J. Colorectal Dis. 2011, 26, 847–858. [Google Scholar] [CrossRef]
- López-Ayllón, B.D.; de Castro-Carpeño, J.; Rodriguez, C.; Pernía, O.; Ibañez de Cáceres, I.; Belda-Iniesta, C.; Perona, R.; Sastre, L. Biomarkers of erlotinib response in non-small cell lung cancer tumors that do not harbor the more common epidermal growth factor receptor mutations. Int. J. Clin. Exp. Pathol. 2015, 8, 2888–2898. [Google Scholar]
- Arcaroli, J.J.; Quackenbush, K.S.; Powell, R.W.; Pitts, T.M.; Spreafico, A.; Varella-Garcia, M.; Bemis, L.; Tan, A.C.; Reinemann, J.M.; Touban, B.M.; et al. Common PIK3CA mutants and a novel 3′ UTR mutation are associated with increased sensitivity to saracatinib. Clin. Cancer Res. 2012, 18, 2704–2714. [Google Scholar] [CrossRef] [Green Version]
- Selga, E.; Morales, C.; Noé, V.; Peinado, M.A.; Ciudad, C.J. Role of caveolin 1, E-cadherin, Enolase 2 and PKCalpha on resistance to methotrexate in human HT29 colon cancer cells. BMC Med. Genom. 2008, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Selga, E.; Oleaga, C.; Ramírez, S.; de Almagro, M.C.; Noé, V.; Ciudad, C.J. Networking of differentially expressed genes in human cancer cells resistant to methotrexate. Genome Med. 2009, 1, 83. [Google Scholar] [CrossRef] [Green Version]
- Mencia, N.; Selga, E.; Noé, V.; Ciudad, C.J. Underexpression of miR-224 in methotrexate resistant human colon cancer cells. Biochem. Pharmacol. 2011, 82, 1572–1582. [Google Scholar] [CrossRef]
- Hua, F.; Shang, S.; Yang, Y.; Zhang, H.; Xu, T.; Yu, J.; Zhou, D.; Cui, B.; Li, K.; Lv, X.; et al. TRIB3 Interacts With β-Catenin and TCF4 to Increase Stem Cell Features of Colorectal Cancer Stem Cells and Tumorigenesis. Gastroenterology 2019, 156, 708–721.e15. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Yang, L.; Shi, H.; Du, W.; Qi, Y.; Qiu, C.; Liang, X.; Shi, W.; Liu, J. Endoplasmic reticulum-targeting photosensitizer Hypericin confers chemo-sensitization towards oxaliplatin through inducing pro-death autophagy. Int. J. Biochem. Cell Biol. 2017, 87, 54–68. [Google Scholar] [CrossRef]
- Tsai, D.-H.; Chung, C.-H.; Lee, K.-T. Antrodia cinnamomea induces autophagic cell death via the CHOP/TRB3/Akt/mTOR pathway in colorectal cancer cells. Sci. Rep. 2018, 8, 17424. [Google Scholar] [CrossRef]
- Hwang, W.-L.; Yang, M.-H.; Tsai, M.-L.; Lan, H.-Y.; Su, S.-H.; Chang, S.-C.; Teng, H.-W.; Yang, S.-H.; Lan, Y.-T.; Chiou, S.-H.; et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 2011, 141, 279–291.e5. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-S.; Lee, C.; Bonifant, C.L.; Ressom, H.; Waldman, T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol. Cell. Biol. 2007, 27, 662–677. [Google Scholar] [CrossRef] [Green Version]
- Yoon, H.; Liyanarachchi, S.; Wright, F.A.; Davuluri, R.; Lockman, J.C.; de la Chapelle, A.; Pellegata, N.S. Gene expression profiling of isogenic cells with different TP53 gene dosage reveals numerous genes that are affected by TP53 dosage and identifies CSPG2 as a direct target of p53. Proc. Natl. Acad. Sci. USA 2002, 99, 15632–15637. [Google Scholar] [CrossRef] [Green Version]
- Mokry, M.; Hatzis, P.; Schuijers, J.; Lansu, N.; Ruzius, F.-P.; Clevers, H.; Cuppen, E. Integrated genome-wide analysis of transcription factor occupancy, RNA polymerase II binding and steady-state RNA levels identify differentially regulated functional gene classes. Nucleic Acids Res. 2012, 40, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theng, S.S.; Wang, W.; Mah, W.-C.; Chan, C.; Zhuo, J.; Gao, Y.; Qin, H.; Lim, L.; Chong, S.S.; Song, J.; et al. Disruption of FAT10-MAD2 binding inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2014, 111, E5282–E5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, K.; Mitter, R.; Muir, M.; Jodrell, D.; Guichard, S. Stable XIAP knockdown clones of HCT116 colon cancer cells are more sensitive to TRAIL, taxanes and irradiation in vitro. Cancer Chemother. Pharmacol. 2009, 64, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katkoori, V.R.; Shanmugam, C.; Jia, X.; Vitta, S.P.; Sthanam, M.; Callens, T.; Messiaen, L.; Chen, D.; Zhang, B.; Bumpers, H.L.; et al. Prognostic significance and gene expression profiles of p53 mutations in microsatellite-stable stage III colorectal adenocarcinomas. PLoS ONE 2012, 7, e30020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Link, W. Tribbles breaking bad: TRIB2 suppresses FOXO and acts as an oncogenic protein in melanoma. Biochem. Soc. Trans. 2015, 43, 1085–1088. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kalveram, B.; Raasi, S.; Groettrup, M.; Schmidtke, G. FAT10, a ubiquitin-independent signal for proteasomal degradation. Mol. Cell. Biol. 2005, 25, 3483–3491. [Google Scholar] [CrossRef] [Green Version]
- Lundemo, A.G.; Pettersen, C.H.; Berge, K.; Berge, R.K.; Schønberg, S.A. Tetradecylthioacetic acid inhibits proliferation of human SW620 colon cancer cells-gene expression profiling implies endoplasmic reticulum stress. Lipids Health Dis. 2011, 10, 190. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.; Sun, H.; Yu, F.-B.; Li, B.; Zhang, Y.; Zhu, Y.-T. The Role of Cyclooxygenase-2 in Colorectal Cancer. Int. J. Med. Sci. 2020, 17, 1095–1101. [Google Scholar] [CrossRef]
- Veettil, S.K.; Nathisuwan, S.; Ching, S.M.; Jinatongthai, P.; Lim, K.G.; Kew, S.T.; Chaiyakunapruk, N. Efficacy and safety of celecoxib on the incidence of recurrent colorectal adenomas: A systematic review and meta-analysis. Cancer Manag. Res. 2019, 11, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grösch, S.; Maier, T.J.; Schiffmann, S.; Geisslinger, G. Cyclooxygenase-2 (COX-2)–Independent Anticarcinogenic Effects of Selective COX-2 Inhibitors. JNCI J. Natl. Cancer Inst. 2006, 98, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Auman, J.T.; Church, R.; Lee, S.-Y.; Watson, M.A.; Fleshman, J.W.; Mcleod, H.L. Celecoxib pre-treatment in human colorectal adenocarcinoma patients is associated with gene expression alterations suggestive of diminished cellular proliferation. Eur. J. Cancer 2008, 44, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Vallinas, M.; Molina, S.; Vicente, G.; Zarza, V.; Martín-Hernández, R.; García-Risco, M.R.; Fornari, T.; Reglero, G.; Ramírez de Molina, A. Expression of microRNA-15b and the glycosyltransferase GCNT3 correlates with antitumor efficacy of Rosemary diterpenes in colon and pancreatic cancer. PLoS ONE 2014, 9, e98556. [Google Scholar] [CrossRef] [Green Version]
- Schoumacher, M.; Hurov, K.E.; Lehár, J.; Yan-Neale, Y.; Mishina, Y.; Sonkin, D.; Korn, J.M.; Flemming, D.; Jones, M.D.; Antonakos, B.; et al. Inhibiting Tankyrases sensitizes KRAS-mutant cancer cells to MEK inhibitors via FGFR2 feedback signaling. Cancer Res. 2014, 74, 3294–3305. [Google Scholar] [CrossRef] [Green Version]
- Berkofsky-Fessler, W.; Nguyen, T.Q.; Delmar, P.; Molnos, J.; Kanwal, C.; DePinto, W.; Rosinski, J.; McLoughlin, P.; Ritland, S.; DeMario, M.; et al. Preclinical biomarkers for a cyclin-dependent kinase inhibitor translate to candidate pharmacodynamic biomarkers in phase I patients. Mol. Cancer Ther. 2009, 8, 2517–2525. [Google Scholar] [CrossRef] [Green Version]
- West, J.D.; Marnett, L.J. Alterations in gene expression induced by the lipid peroxidation product, 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 2005, 18, 1642–1653. [Google Scholar] [CrossRef]
- Luesch, H.; Chanda, S.K.; Raya, R.M.; DeJesus, P.D.; Orth, A.P.; Walker, J.R.; Izpisúa Belmonte, J.C.; Schultz, P.G. A functional genomics approach to the mode of action of apratoxin A. Nat. Chem. Biol. 2006, 2, 158–167. [Google Scholar] [CrossRef]
- Gasparovic, A.C.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. Cancer growth regulation by 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 226–234. [Google Scholar] [CrossRef]
- Wu, J.; Tao, W.-W.; Chong, D.-Y.; Lai, S.-S.; Wang, C.; Liu, Q.; Zhang, T.-Y.; Xue, B.; Li, C.-J. Early growth response-1 negative feedback regulates skeletal muscle postprandial insulin sensitivity via activating Ptp1b transcription. FASEB J. 2018, 32, 4370–4379. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Takatsuno, Y.; Kim, H.; Hirose, H.; Sekimoto, M.; Doki, Y.; Mori, M. Abnormal expression of TRIB3 in colorectal cancer: A novel marker for prognosis. Br. J. Cancer 2009, 101, 1664–1670. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.; Yalla, K.; Salomé, M.; Moka, H.A.; Castañeda, E.G.; Eyers, P.A.; Keeshan, K. Trib2 expression in granulocyte-monocyte progenitors drives a highly drug resistant acute myeloid leukaemia linked to elevated Bcl2. Oncotarget 2018, 9, 14977–14992. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zuo, J.; Wahafu, A.; Wang, M.; Li, R.-C.; Xie, W.-F. Combined elevation of TRIB2 and MAP3K1 indicates poor prognosis and chemoresistance to temozolomide in glioblastoma. CNS Neurosci. Ther. 2020, 26, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Petiwala, S.M.; Johnson, J.J. Diterpenes from rosemary (Rosmarinus officinalis): Defining their potential for anti-cancer activity. Cancer Lett. 2015, 367, 93–102. [Google Scholar] [CrossRef]
- Moore, J.; Yousef, M.; Tsiani, E. Anticancer Effects of Rosemary (Rosmarinus officinalis L.) Extract and Rosemary Extract Polyphenols. Nutrients 2016, 8, 731. [Google Scholar] [CrossRef]
- Valdés, A.; Sullini, G.; Ibáñez, E.; Cifuentes, A.; García-Cañas, V. Rosemary polyphenols induce unfolded protein response and changes in cholesterol metabolism in colon cancer cells. J. Funct. Foods 2015, 15, 429–439. [Google Scholar] [CrossRef]
- Valdés, A.; Artemenko, K.A.; Bergquist, J.; García-Cañas, V.; Cifuentes, A. Comprehensive Proteomic Study of the Antiproliferative Activity of a Polyphenol-Enriched Rosemary Extract on Colon Cancer Cells Using Nanoliquid Chromatography–Orbitrap MS/MS. J. Proteome Res. 2016, 15, 1971–1985. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Li, G.; Petiwala, S.M.; Householter, E.; Johnson, J.J. Standardized rosemary (Rosmarinus officinalis) extract induces Nrf2/sestrin-2 pathway in colon cancer cells. J. Funct. Foods 2015, 13, 137–147. [Google Scholar] [CrossRef]
- Hill, R.; Madureira, P.A.; Ferreira, B.; Baptista, I.; Machado, S.; Colaço, L.; dos Santos, M.; Liu, N.; Dopazo, A.; Ugurel, S.; et al. TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat. Commun. 2017, 8, 14687. [Google Scholar] [CrossRef]
- Moore, J.; Megaly, M.; MacNeil, A.J.; Klentrou, P.; Tsiani, E. Rosemary extract reduces Akt/mTOR/p70S6K activation and inhibits proliferation and survival of A549 human lung cancer cells. Biomed. Pharmacother. 2016, 83, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. 4-Hydroxy-2-nonenal: A product and mediator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Ji, C.; Amarnath, V.; Pietenpol, J.A.; Marnett, L.J. 4-hydroxynonenal induces apoptosis via caspase-3 activation and cytochrome c release. Chem. Res. Toxicol. 2001, 14, 1090–1096. [Google Scholar] [CrossRef]
- Bao, X.-Y.; Sun, M.; Peng, T.-T.; Han, D.-M. TRIB3 promotes proliferation, migration, and invasion of retinoblastoma cells by activating the AKT/mTOR signaling pathway. Cancer Biomark. 2021, 31, 307–315. [Google Scholar] [CrossRef]
- Shen, P.; Zhang, T.-Y.; Wang, S.-Y. TRIB3 promotes oral squamous cell carcinoma cell proliferation by activating the AKT signaling pathway. Exp. Ther. Med. 2021, 21, 313. [Google Scholar] [CrossRef]
- Wennemers, M.; Stegeman, H.; Bussink, J.; Versleijen-Jonkers, Y.M.H.; van Laarhoven, H.W.M.; Raleigh, J.A.; Varia, M.A.; Sweep, F.C.G.J.; Span, P.N. Hypoxia regulation of phosphokinases and the prognostic value of pAKT in breast cancer. Int. J. Biol. Markers 2013, 28, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Liu, B.; Li, B.; Du, G.; Li, Y.; Wang, J.; He, L.; Wan, X. TRIB3 suppresses proliferation and invasion and promotes apoptosis of endometrial cancer cells by regulating the AKT signaling pathway. OncoTargets Ther. 2019, 12, 2235–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, M.; Lorente, M.; García-Taboada, E.; Gómez, E.P.; Dávila, D.; Zúñiga-García, P.; Flores, J.M.; Rodríguez, A.; Hegedus, Z.; Mosén-Ansorena, D.; et al. TRIB3 suppresses tumorigenesis by controlling mTORC2/AKT/FOXO signaling. Mol. Cell. Oncol. 2015, 2, e980134. [Google Scholar] [CrossRef]
- Salazar, M.; Lorente, M.; García-Taboada, E.; Pérez Gómez, E.; Dávila, D.; Zúñiga-García, P.; María Flores, J.; Rodríguez, A.; Hegedus, Z.; Mosén-Ansorena, D.; et al. Loss of Tribbles pseudokinase-3 promotes Akt-driven tumorigenesis via FOXO inactivation. Cell Death Differ. 2015, 22, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.G.L.; Ren, J.; Cheong, I.S.Y.; Ban, K.H.K.; Ooi, L.L.P.J.; Yong Tan, S.; Kan, A.; Nuchprayoon, I.; Jin, R.; Lee, K.-H.; et al. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene 2003, 22, 2592–2603. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Theng, S.S.; Zhuo, J.; Teo, W.B.; Ren, J.; Lee, C.G.L. FAT10, an ubiquitin-like protein, confers malignant properties in non-tumorigenic and tumorigenic cells. Carcinogenesis 2014, 35, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Dong, Z.; Liang, J.; Cao, C.; Sun, J.; Ding, Y.; Wu, D. As an independent prognostic factor, FAT10 promotes hepatitis B virus-related hepatocellular carcinoma progression via Akt/GSK3β pathway. Oncogene 2014, 33, 909–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Du, Y.; Cheng, C.; Deng, X.; Shi, Z.; Lu, X.; Hu, H.; Qiu, J.; Jiang, W. FAT10 promotes the progression of bladder cancer by upregulating HK2 through the EGFR/AKT pathway. Exp. Cell Res. 2021, 398, 112401. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, M.T.; Yassuda, V.; Bragança, J.; Link, W.; Ferreira, B.I.; De Sousa-Coelho, A.L. Tribbles Gene Expression Profiles in Colorectal Cancer. Gastrointest. Disord. 2021, 3, 218-236. https://doi.org/10.3390/gidisord3040021
Fernandes MT, Yassuda V, Bragança J, Link W, Ferreira BI, De Sousa-Coelho AL. Tribbles Gene Expression Profiles in Colorectal Cancer. Gastrointestinal Disorders. 2021; 3(4):218-236. https://doi.org/10.3390/gidisord3040021
Chicago/Turabian StyleFernandes, Mónica T., Victor Yassuda, José Bragança, Wolfgang Link, Bibiana I. Ferreira, and Ana Luísa De Sousa-Coelho. 2021. "Tribbles Gene Expression Profiles in Colorectal Cancer" Gastrointestinal Disorders 3, no. 4: 218-236. https://doi.org/10.3390/gidisord3040021
APA StyleFernandes, M. T., Yassuda, V., Bragança, J., Link, W., Ferreira, B. I., & De Sousa-Coelho, A. L. (2021). Tribbles Gene Expression Profiles in Colorectal Cancer. Gastrointestinal Disorders, 3(4), 218-236. https://doi.org/10.3390/gidisord3040021