Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review




Abstract
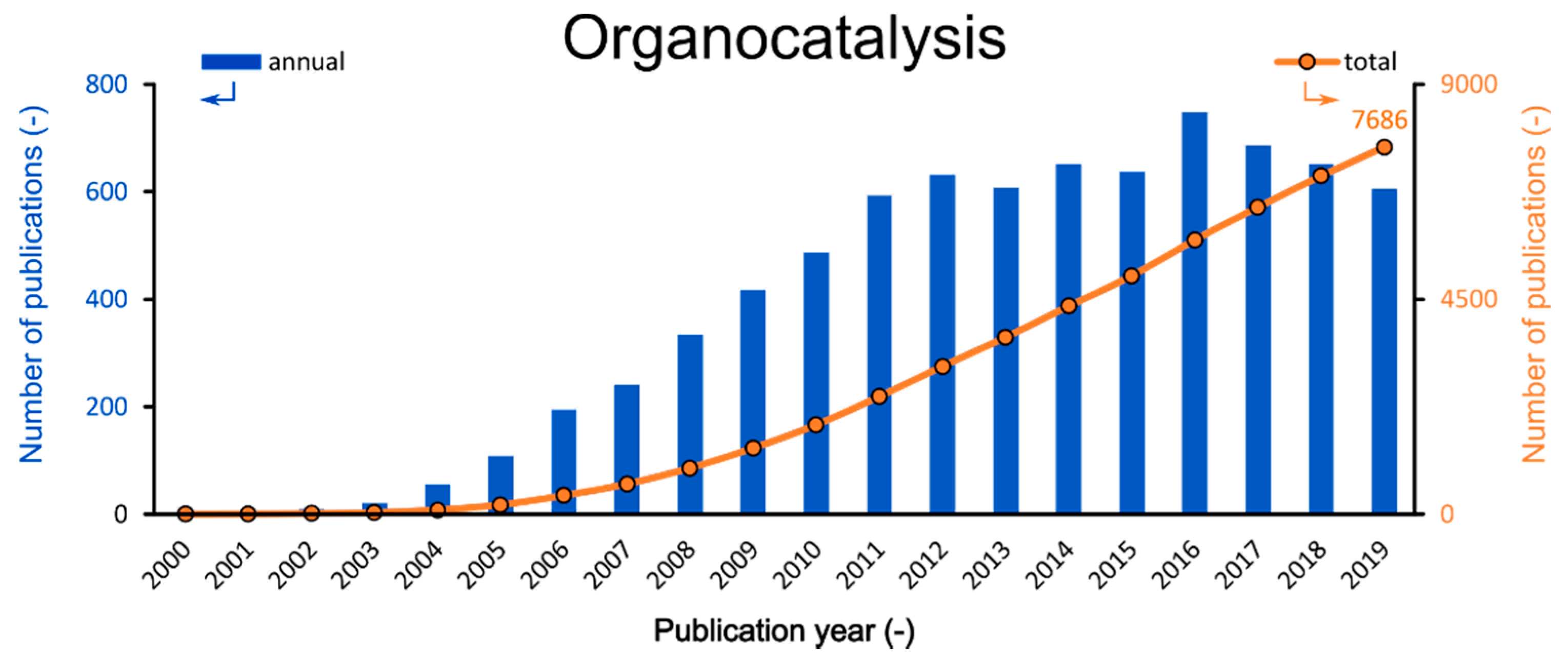
:1. Introduction
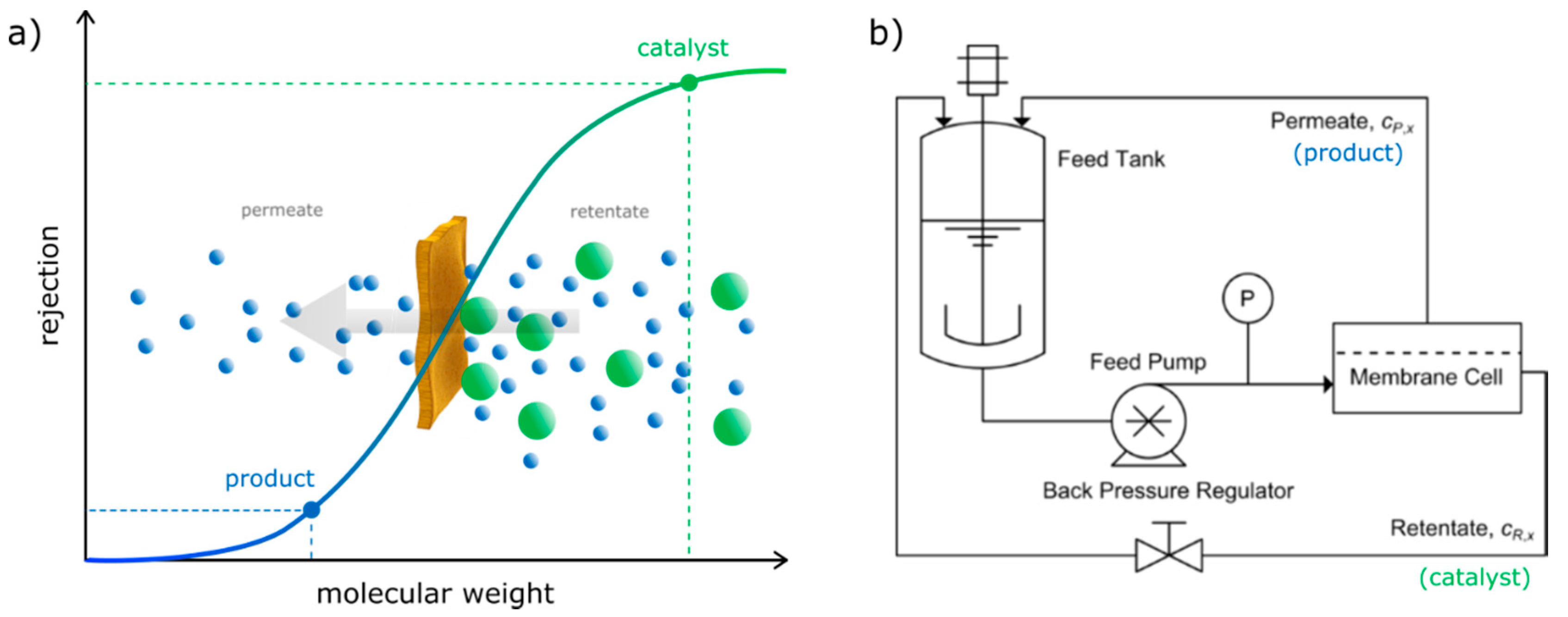
2. Homogeneous Organocatalyst Recovery Using Organic Solvent Nanofiltration
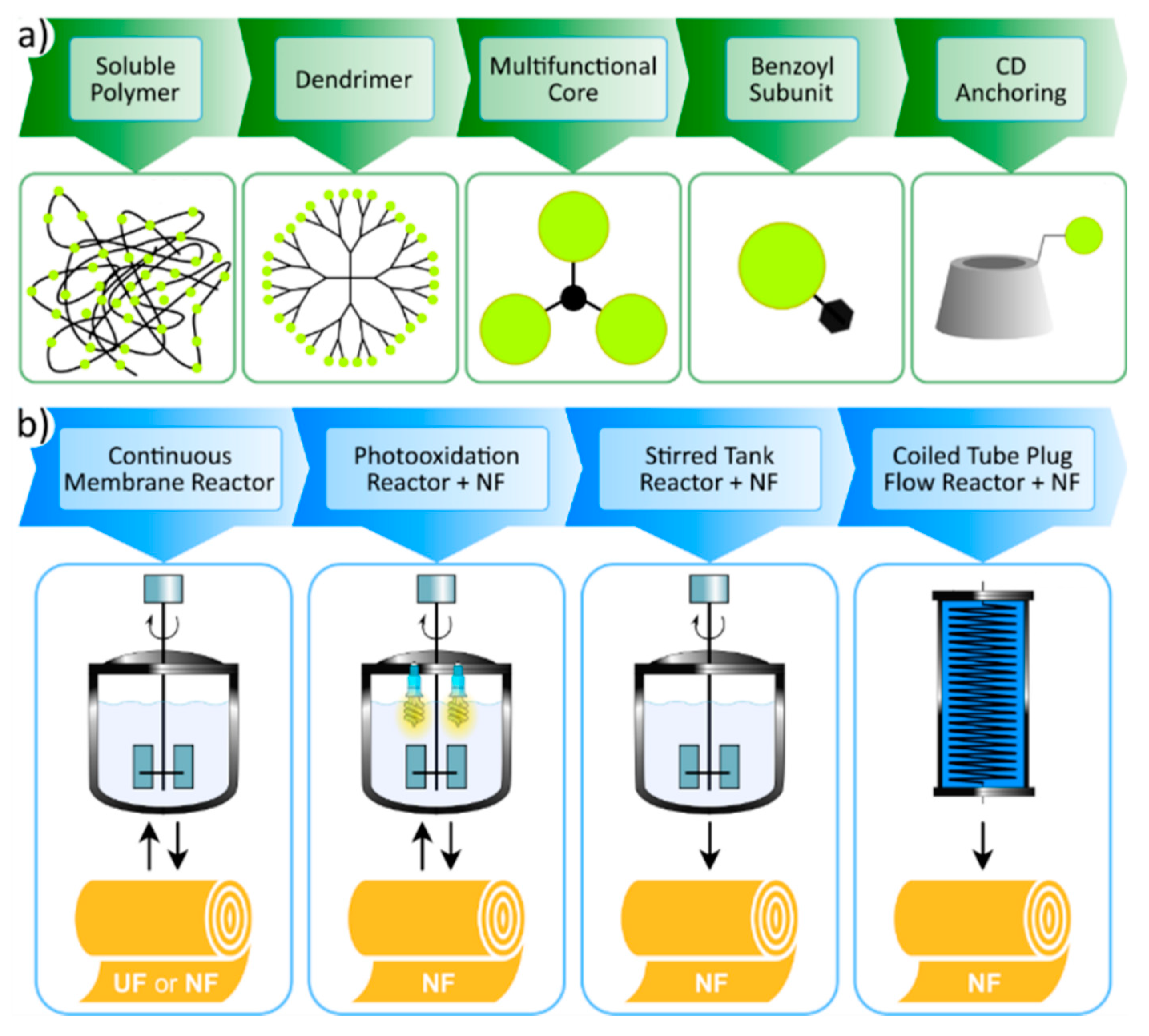
3. Molecular Weight Enlargement of Homogeneous Organocatalysts for Membrane Filtration
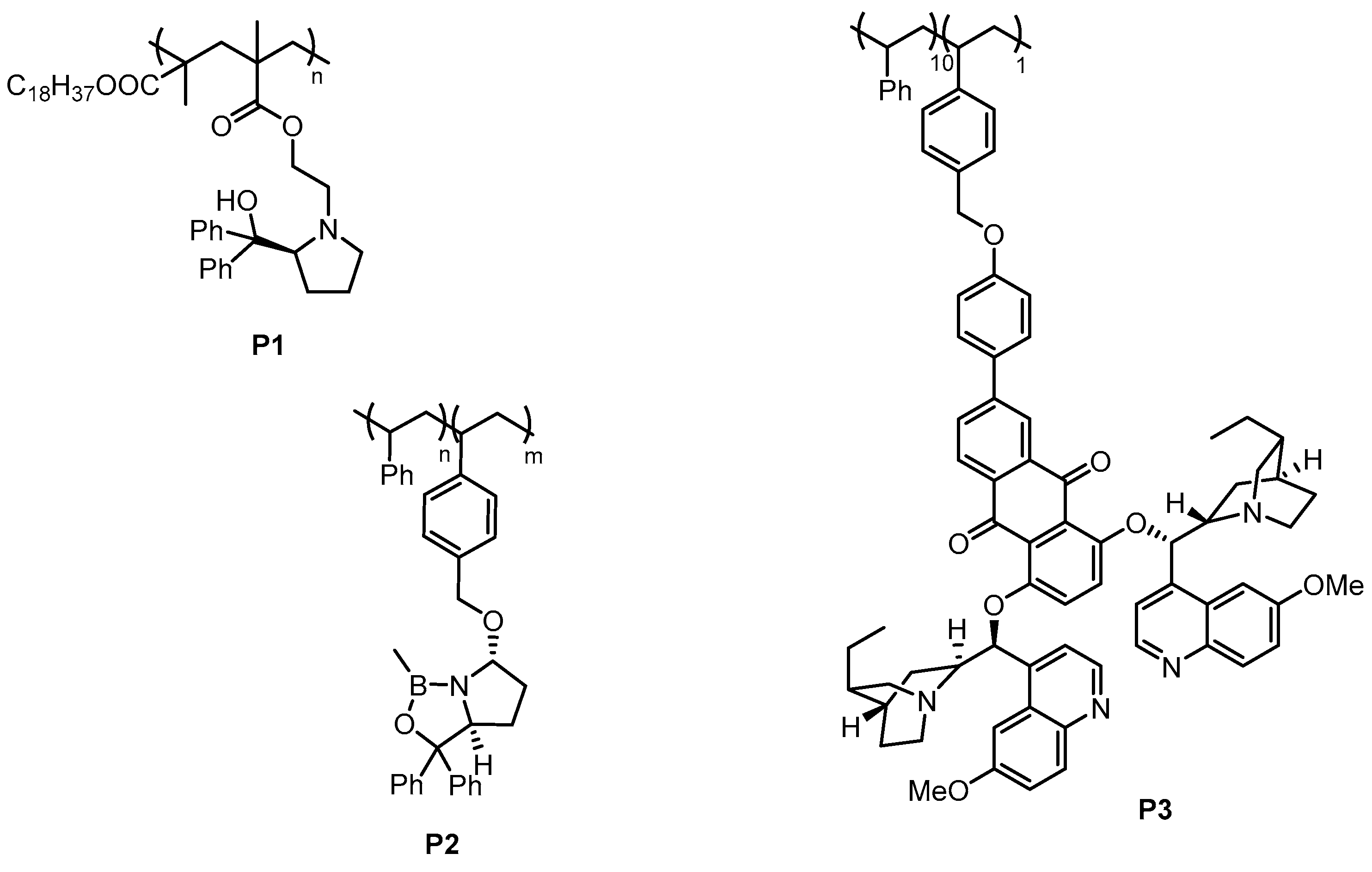
3.1. Soluble Polymers
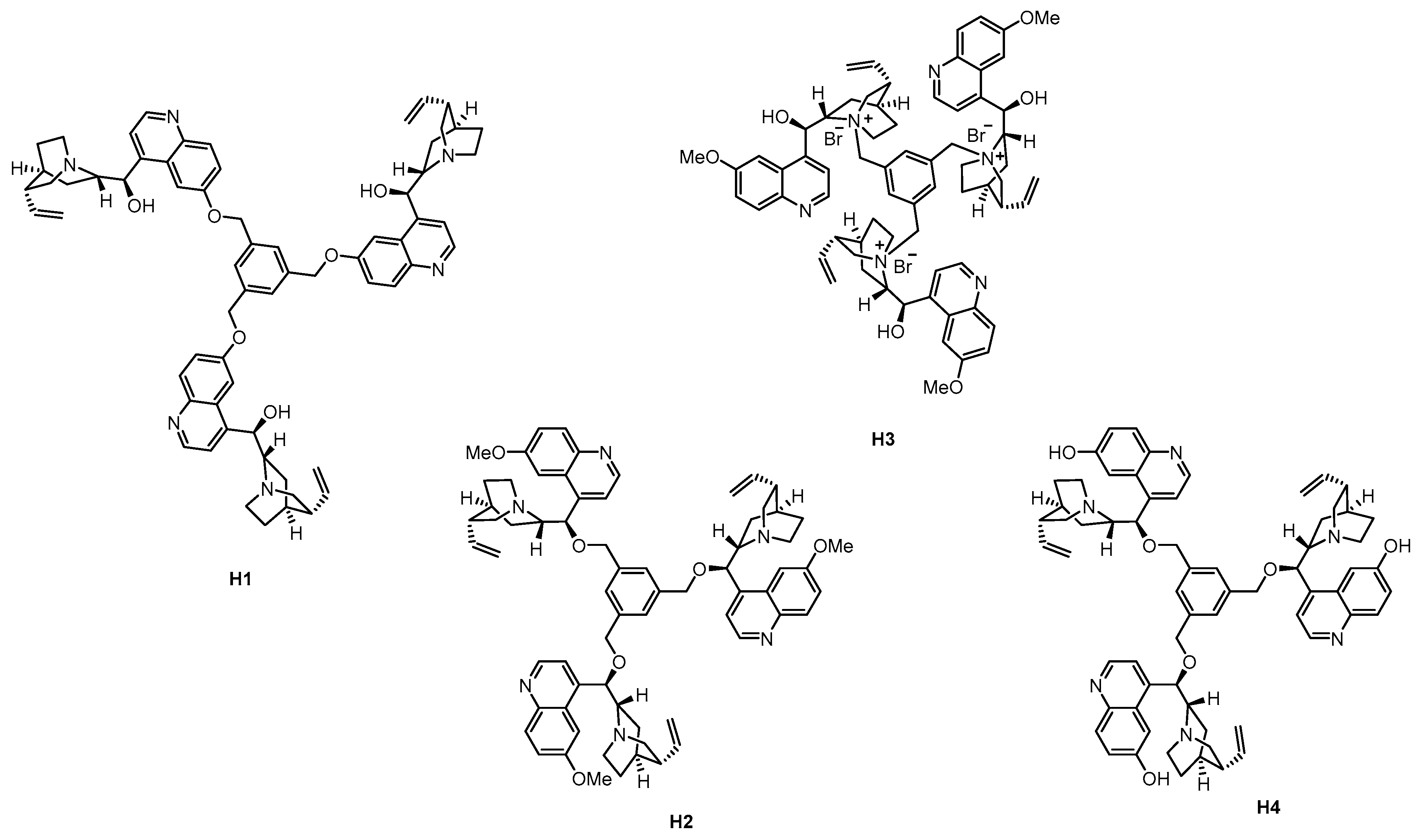
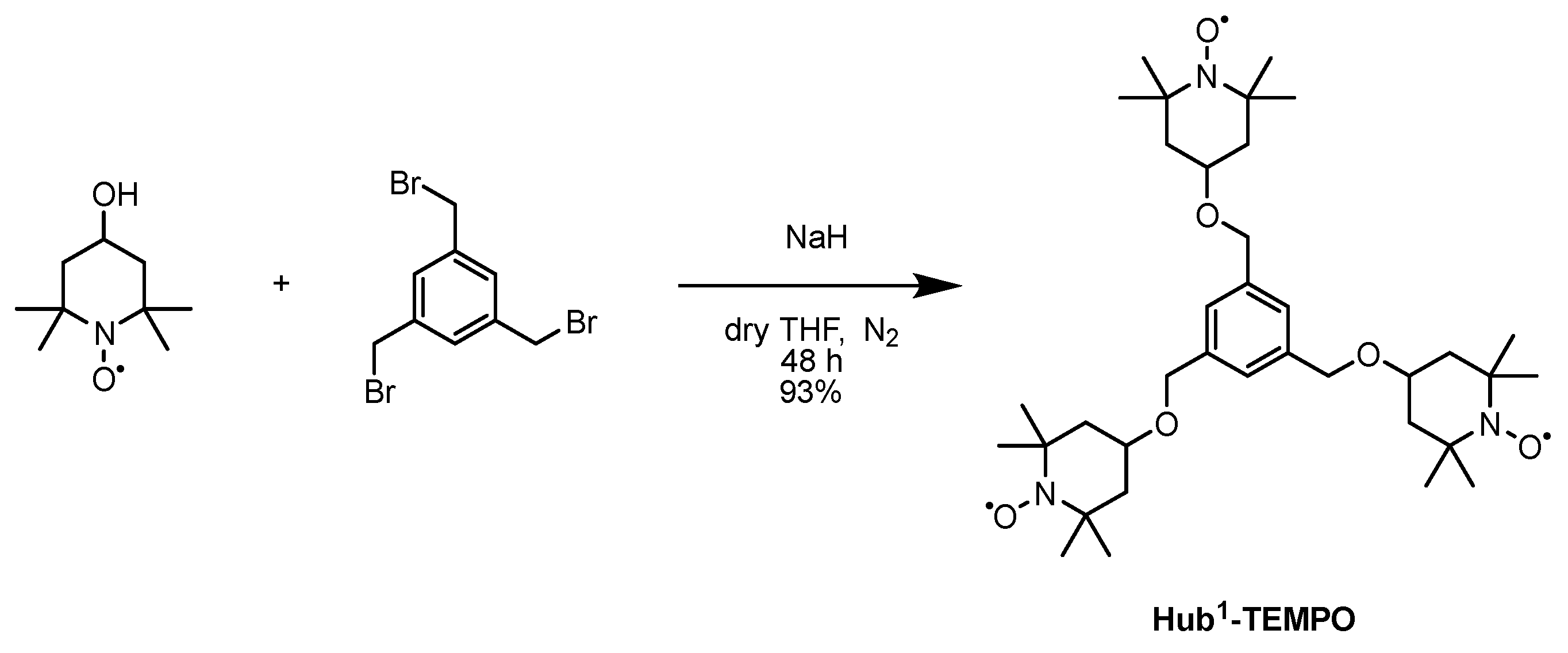
3.2. Dendrimers
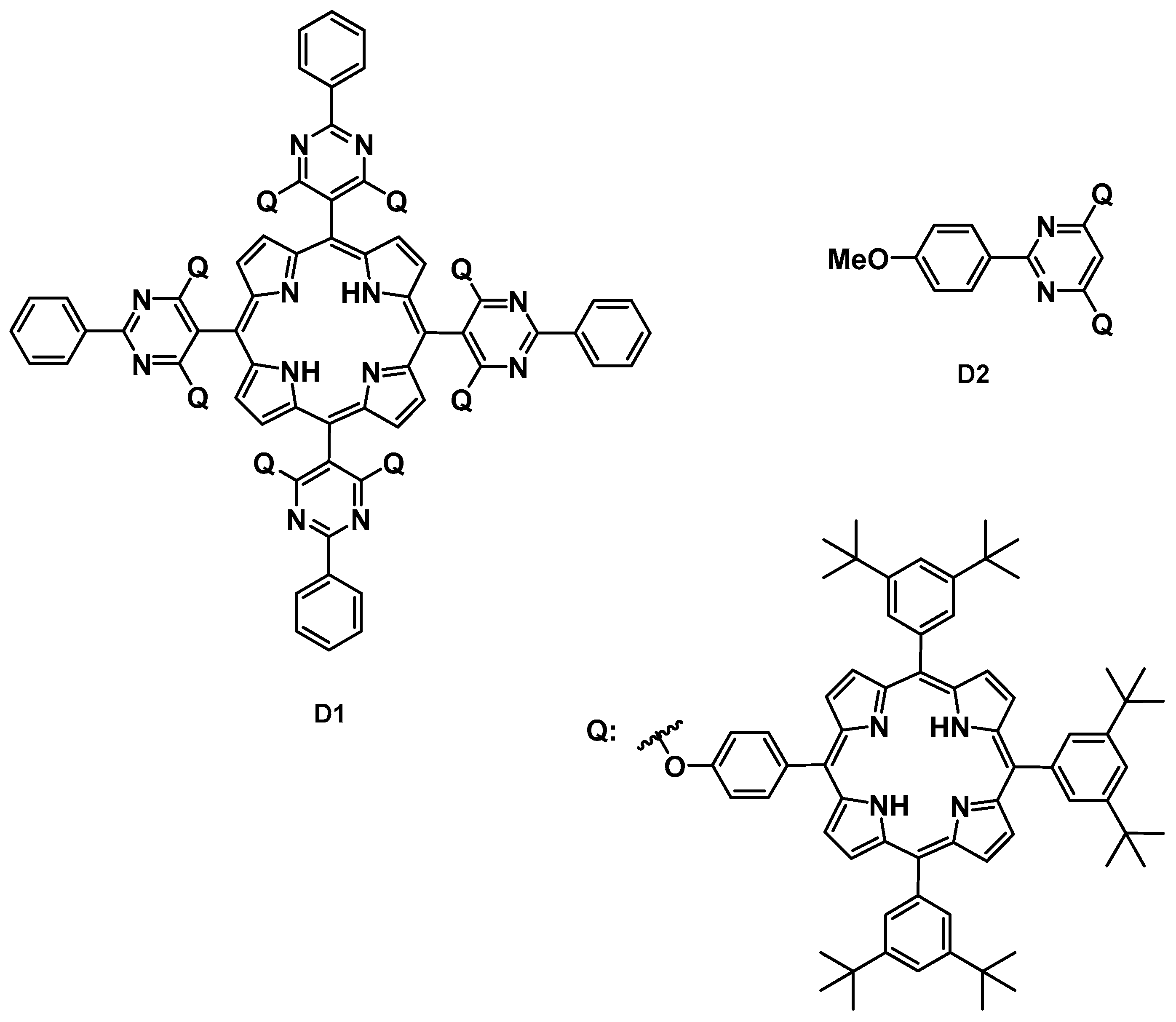
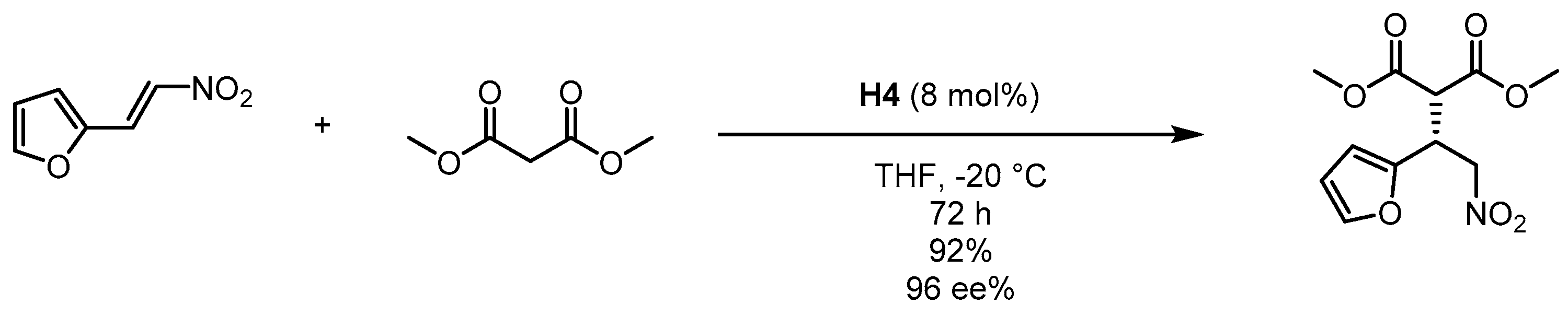
3.3. Multifunctional Core
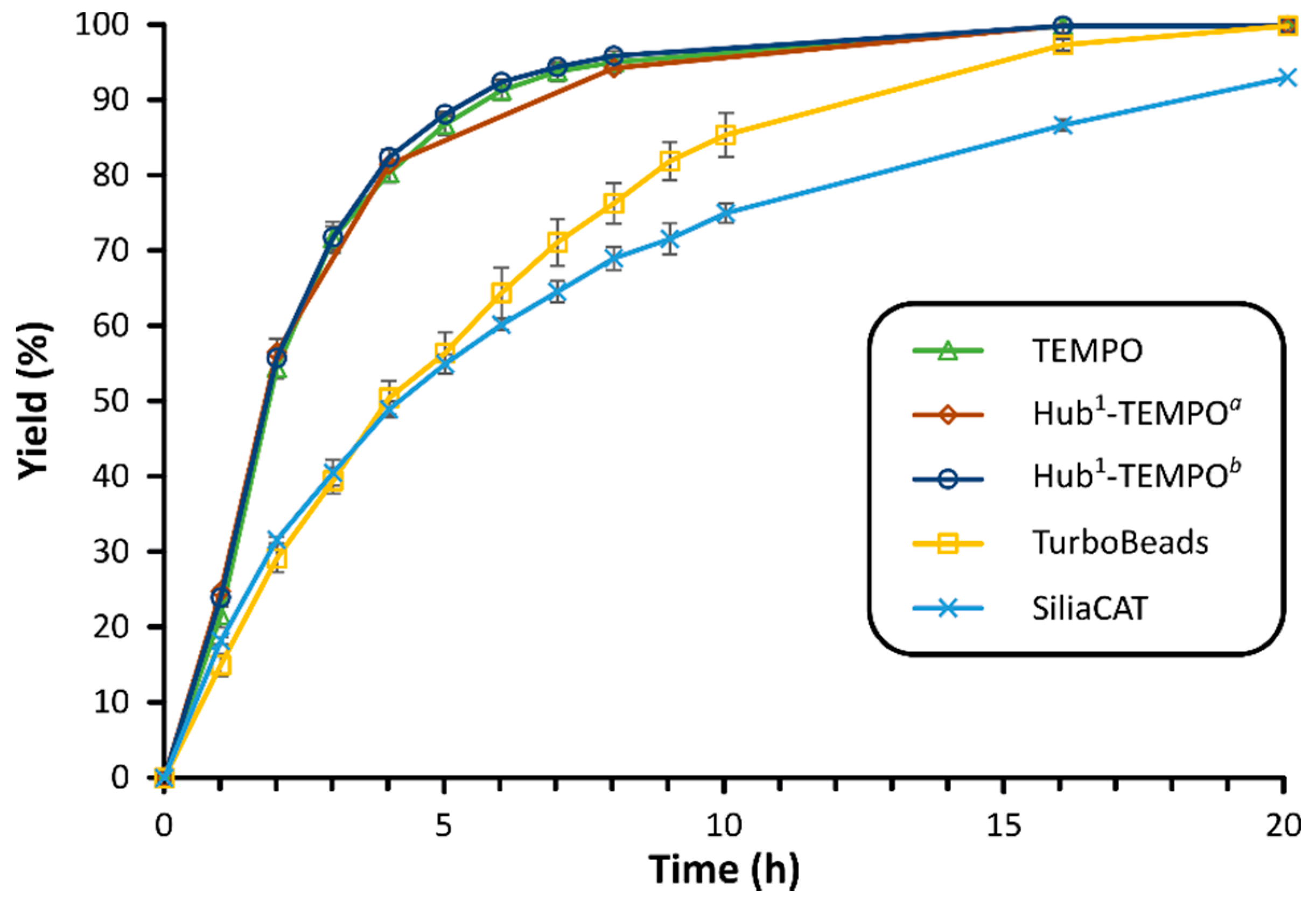
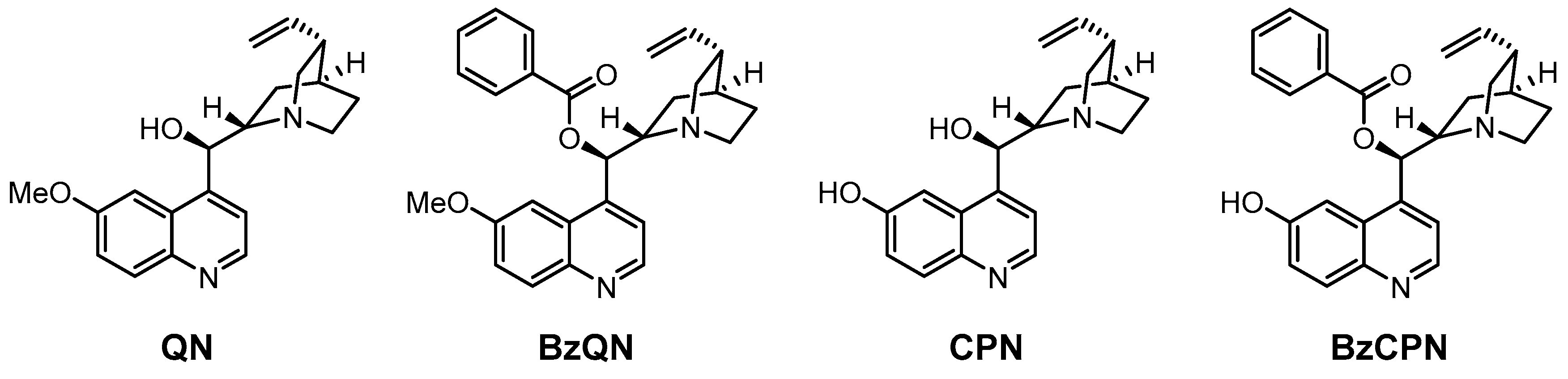
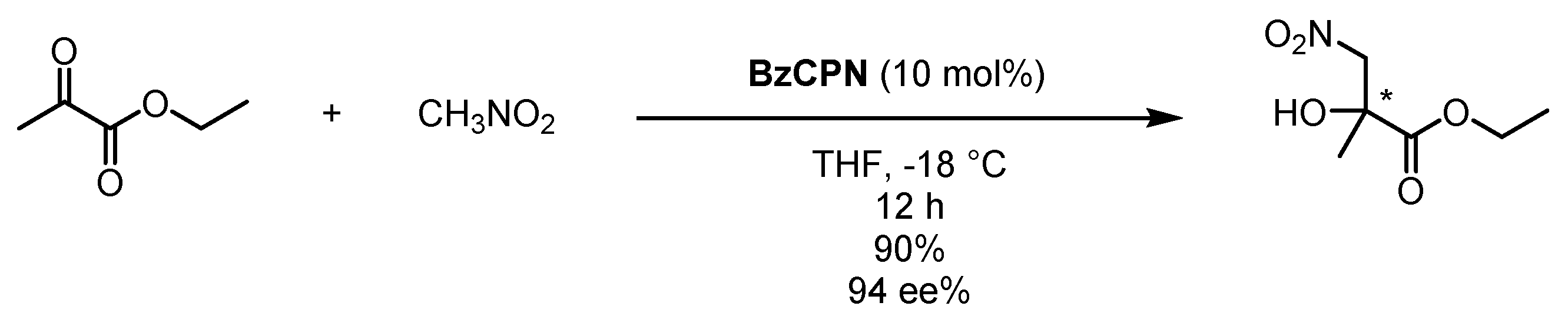
3.4. Benzoyl Subunit
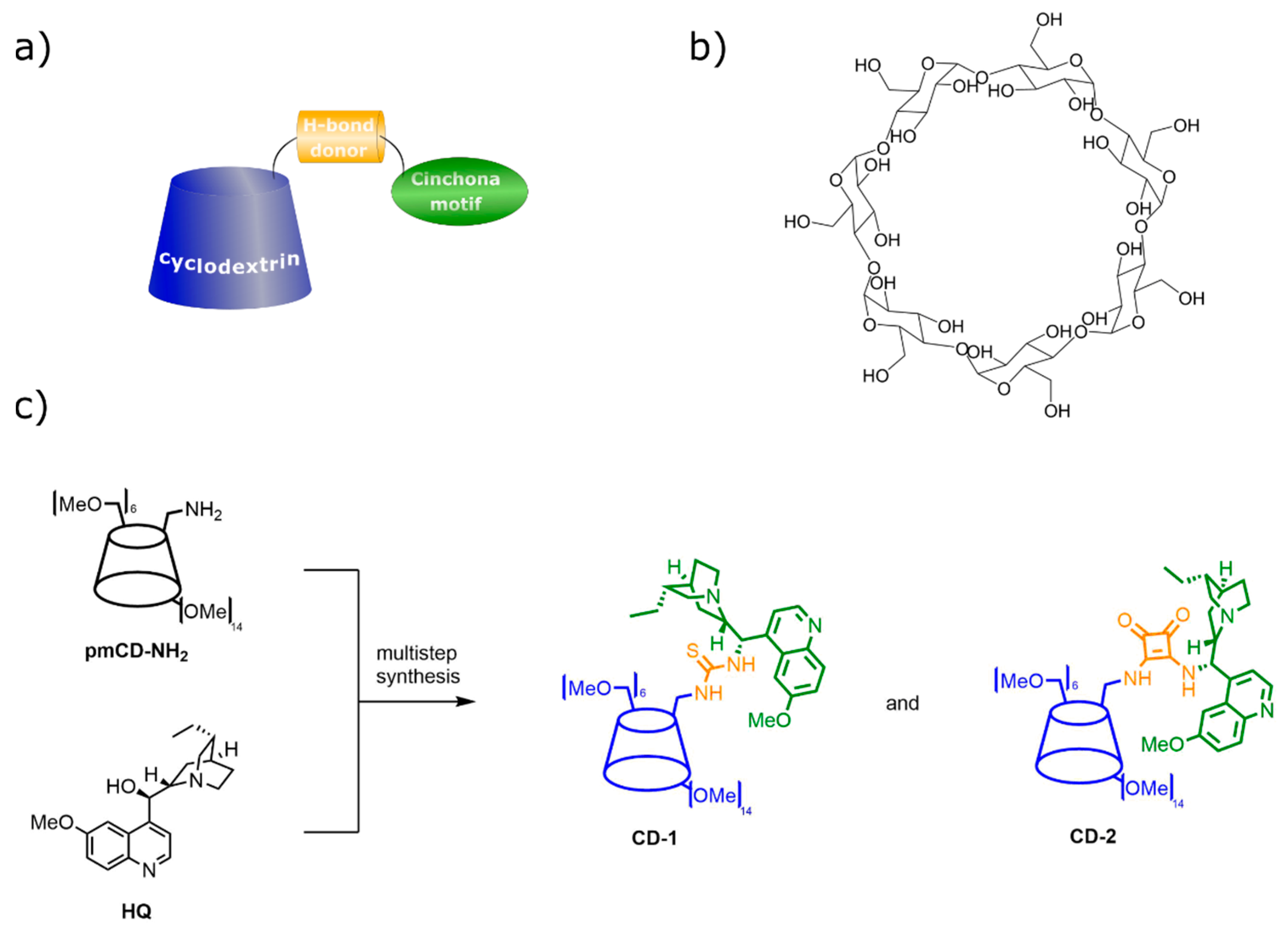
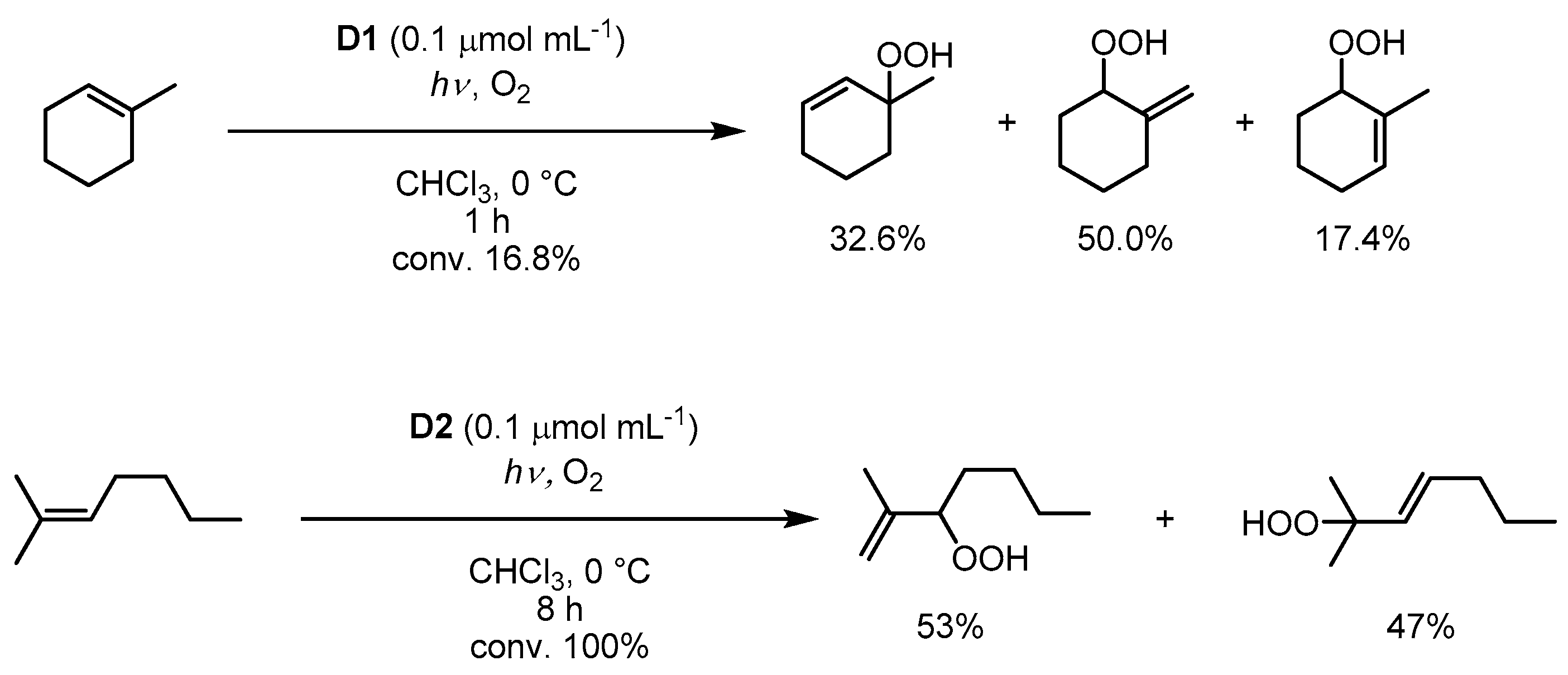
3.5. Cyclodextrin Anchoring
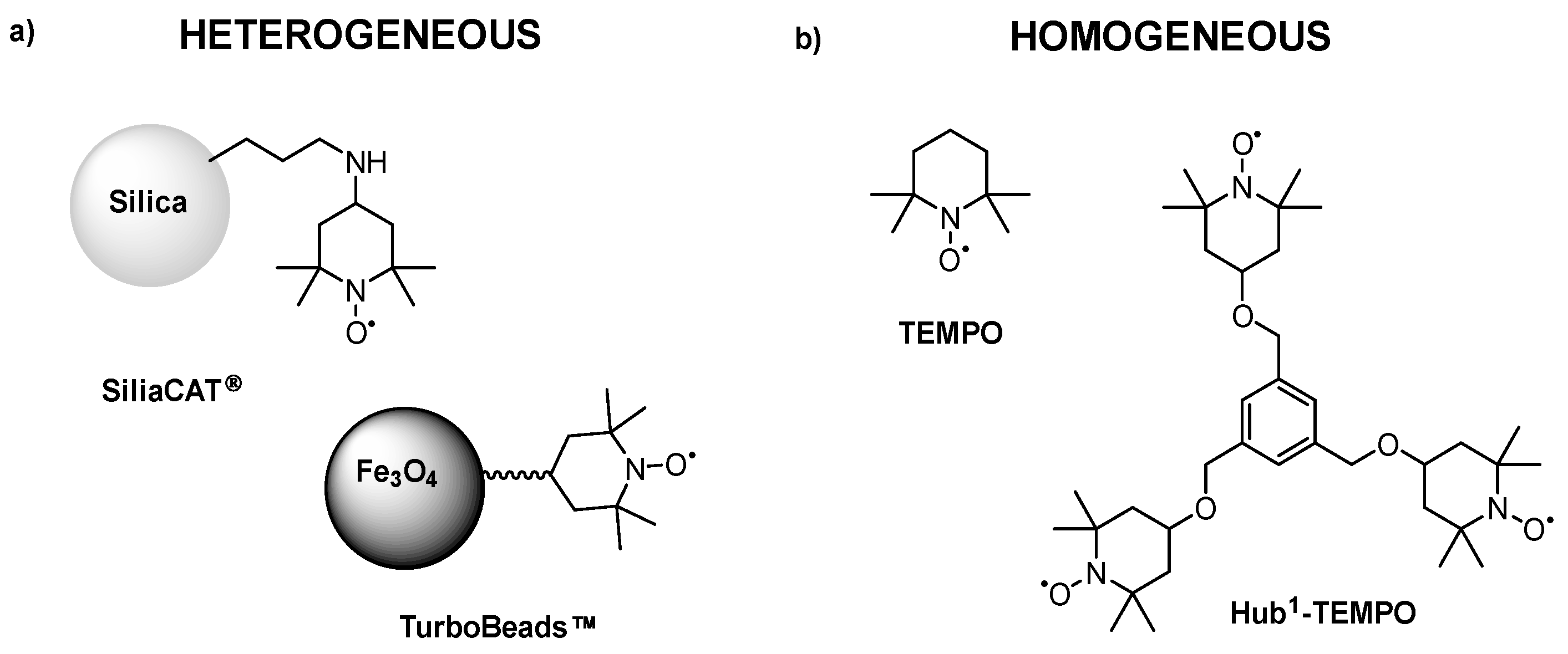
3.6. Explored Membrane-Processes
4. Conclusions and Outlook
Funding

Conflicts of Interest
References
- Catalyst Market by Material, Type, Application, Regions, Industry Analysis, Size, Share, Growth, Trends, and Forecast 2018 to 2025. Available online: https://www.fiormarkets.com (accessed on 18 March 2020).
- Kamer, P.; Vogt, D.; Thybaut, J.W. Contemporary Catalysis: Science, Technology and Applications; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen—Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef]
- Akiyama, T. Stronger Brønsted Acids. Chem. Rev. 2007, 107, 5744–5758. [Google Scholar] [CrossRef]
- Enders, D.; Grondal, C.; Hüttl, M.R.M. Asymmetric Organocatalytic Domino Reactions. Angew. Chem. Int. Ed. 2007, 46, 1570–1581. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The Advent and Development of Organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- Oliveira, V.; Cardoso, M.; Forezi, L. Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef] [Green Version]
- Carlone, A.; Bernardi, L. Enantioselective Organocatalytic Approaches to Active Pharmaceutical Ingredients – Selected Industrial Examples. Phys. Sci. Rev. 2019, 4, 20180097. [Google Scholar] [CrossRef]
- Transforming Our World: The 2030 Agenda for Sustainable Development: Sustainable Development Knowledge Platform. Available online: https://sustainabledevelopment.un.org/post2015/transformingourworld/publication (accessed on 18 March 2020).
- Sheldon, R.A.; Arends, I.; Hanefeld, U. Green Chemistry and Catalysis; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Livingston, A.G.; Trout, B.L.; Horvath, I.T.; Johnson, M.D.; Vaccaro, L.; Coronas, J.; Babbitt, C.W.; Zhang, X.; Pradeep, T.; Drioli, E.; et al. Challenges and Directions for Green Chemical Engineering—Role of Nanoscale Materials. In Sustainable Nanoscale Engineering; Szekely, G., Livingston, A.G., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–18. [Google Scholar]
- Joshi, S.S.; Ranade, V.V. Industrial Catalytic Processes for Fine and Specialty Chemicals; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Mika, L.T.; Horváth, I.T. Fluorous Catalysis. In Green Techniques for Organic Synthesis and Medicinal Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2018; Volume 79, pp. 219–268. [Google Scholar]
- Benaglia, M. Recoverable Organic Catalysts. In Recoverable and Recyclable Catalysts; John Wiley & Sons, Ltd.: Chichester, UK, 2009; pp. 301–340. [Google Scholar]
- Kasaplar, P.; Riente, P.; Hartmann, C.; Pericàs, M.A. A Polystyrene-Supported, Highly Recyclable Squaramide Organocatalyst for the Enantioselective Michael Addition of 1,3-Dicarbonyl Compounds to β-Nitrostyrenes. Adv. Synth. Catal. 2012, 354, 2905–2910. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Y.; Yu, P.; Han, X.; He, J. Exploration of Dependence of Organo-Catalyzed Enantioselective Michael Addition on the Pore Size of Mesoporous Host. ACS Catal. 2012, 2, 1118–1126. [Google Scholar] [CrossRef]
- Rase, H.F. Handbook of Commercial Catalysts; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Nagy, S.; Fehér, Z.; Kárpáti, L.; Bagi, P.; Kisszékelyi, P.; Koczka, B.; Huszthy, P.; Pukánszky, B.; Kupai, J. Synthesis and applications of cinchona squaramide-modified poly(glycidyl methacrylate) microspheres as recyclable polymer-grafted enantioselective organocatalysts. Chem. Eur. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B.H.; Ghorai, S. Organocatalysis in Water at Room Temperature with In-Flask Catalyst Recycling. Org. Lett. 2012, 14, 422–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Großeheilmann, J.; Vanderveen, J.R.; Jessop, P.G.; Kragl, U. Switchable-Hydrophilicity Solvents for Product Isolation and Catalyst Recycling in Organocatalysis. ChemSusChem 2016, 9, 696–702. [Google Scholar] [CrossRef] [Green Version]
- Szekely, G.; Jimenez-Solomon, M.F.; Marchetti, P.; Kim, J.F.; Livingston, A.G. Sustainability Assessment of Organic Solvent Nanofiltration: From Fabrication to Application. Green Chem. 2014, 16, 4440–4473. [Google Scholar] [CrossRef] [Green Version]
- Cseri, L.; Fodi, T.; Kupai, J.; Balogh, G.T.; Garforth, A.; Szekely, G. Membrane-Assisted Catalysis in Organic Media. Adv. Mater. Lett. 2017, 8, 1094–1124. [Google Scholar] [CrossRef] [Green Version]
- Branco, L.C.; Phillips, F.A.M.; Marques, M.M.; Gago, S.; Branco, P.S. Recent Advances in Sustainable Organocatalysis. In Recent Advances in Organocatalysis; Karamé, I., Srour, H., Eds.; InTech: London, UK, 2016. [Google Scholar]
- Krištofíková, D.; Modrocká, V.; Mečiarová, M.; Šebesta, R. Green Asymmetric Organocatalysis. ChemSusChem 2020, 13, 2828–2858. [Google Scholar] [CrossRef]
- Kim, J.F.; Székely, G.; Valtcheva, I.B.; Livingston, A.G. Increasing the Sustainability of Membrane Processes through Cascade Approach and Solvent Recovery—Pharmaceutical Purification Case Study. Green Chem. 2014, 16, 133–145. [Google Scholar] [CrossRef]
- Phuong, H.A.L.; Ayob, N.A.I.; Blanford, C.F.; Rawi, N.F.M.; Szekely, G. Nonwoven Membrane Supports from Renewable Resources: Bamboo Fiber Reinforced Poly(Lactic Acid) Composites. ACS Sustain. Chem. Eng. 2019, 7, 11885–11893. [Google Scholar] [CrossRef]
- Cheng, X.Q.; Wang, Z.X.; Jiang, X.; Li, T.; Lau, C.; Guo, Z.; Ma, J.; Shao, L. Towards Sustainable Ultrafast Molecular-Separation Membranes: From Conventional Polymers to Emerging Materials. Prog. Mater. Sci. 2018, 92, 258–283. [Google Scholar] [CrossRef] [Green Version]
- Nunes, S.P.; Culfaz-Emecen, P.Z.; Ramon, G.Z.; Visser, T.; Koops, G.H.; Jin, W.; Ulbricht, M. Thinking the Future of Membranes: Perspectives for Advanced and New Membrane Materials and Manufacturing Processes. J. Membr. Sci. 2020, 598, 117761. [Google Scholar] [CrossRef]
- Marchetti, P.; Peeva, L.; Livingston, A.G. The Selectivity Challenge in Organic Solvent Nanofiltration: Membrane and Process Solutions. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Galizia, M.; Bye, K.P. Advances in Organic Solvent Nanofiltration Rely on Physical Chemistry and Polymer Chemistry. Front. Chem. 2018, 6, 511. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Lyu, Z.; Gu, Q.; Zhang, L.; Wang, J. Ceramic-Based Membranes for Water and Wastewater Treatment. Colloids Surf. A Physicochem. Eng. Asp. 2019, 578, 123513. [Google Scholar] [CrossRef]
- Schäfer, A.I.; Fane, A.G.; Waite, T.D. Nanofiltration of Natural Organic Matter: Removal, Fouling and the Influence of Multivalent Ions. Desalination 1998, 118, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Košutić, K.; Kaštelan-Kunst, L.; Kunst, B. Porosity of Some Commercial Reverse Osmosis and Nanofiltration Polyamide Thin-Film Composite Membranes. J. Membr. Sci. 2000, 168, 101–108. [Google Scholar] [CrossRef]
- Xu, Y.; Lebrun, R.E. Investigation of the Solute Separation by Charged Nanofiltration Membrane: Effect of pH, Ionic Strength and Solute Type. J. Membr. Sci. 1999, 158, 93–104. [Google Scholar] [CrossRef]
- Van Der Bruggen, B.; Daems, B.; Wilms, D.; Vandecasteele, C. Mechanisms of Retention and Flux Decline for the Nanofiltration of Dye Baths from the Textile Industry. Sep. Purif. Technol. 2001, 22, 519–528. [Google Scholar] [CrossRef]
- Machado, D.R.; Hasson, D.; Semiat, R. Effect of Solvent Properties on Permeate Flow through Nanofiltration Membranes. Part I: Investigation of Parameters Affecting Solvent Flux. J. Membr. Sci. 1999, 163, 93–102. [Google Scholar] [CrossRef]
- Machado, D.R.; Hasson, D.; Semiat, R. Effect of Solvent Properties on Permeate Flow through Nanofiltration Membranes. Part II. Transport Model. J. Membr. Sci. 2000, 166, 63–69. [Google Scholar] [CrossRef]
- Marchetti, P.; Jimenez-Solomon, M.F.; Szekely, G.; Livingston, A.G. Molecular Separation with Organic Solvent Nanofiltration: A Critical Review. Chem. Rev. 2014, 114, 10735–10806. [Google Scholar] [CrossRef] [PubMed]
- Dreimann, J.; Lutze, P.; Zagajewski, M.; Behr, A.; Górak, A.; Vorholt, A.J. Highly Integrated Reactor-Separator Systems for the Recycling of Homogeneous Catalysts. Chem. Eng. Process. Process Intensif. 2016, 99, 124–131. [Google Scholar] [CrossRef]
- Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. Membrane-Grafted Asymmetric Organocatalyst for an Integrated Synthesis-Separation Platform. ACS Catal. 2018, 8, 7430–7438. [Google Scholar] [CrossRef]
- Gürsel, V.I.; Noël, T.; Wang, Q.; Hessel, V. Separation/Recycling Methods for Homogeneous Transition Metal Catalysts in Continuous Flow. Green Chem. 2015, 17, 2012–2026. [Google Scholar] [CrossRef]
- Kisszékelyi, P.; Nagy, S.; Tóth, B.; Zeller, B.; Hegedűs, L.; Mátravölgyi, B.; Grün, A.; Németh, T.; Huszthy, P.; Kupai, J. Synthesis and recovery of pyridine- and piperidine-based camphorsulfonamide organocatalysts used for Michael addition reaction. Period. Polytech. Chem. Eng. 2018, 62, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Kupai, J.; Kisszékelyi, P.; Rojik, E.; Dargó, G.; Hegedűs, L.; Bezzegh, D.; Maszler, P.; Szabó, L.; Németh, T.; Balogh, G.T.; et al. Synthesis and determination of pKa values of new enantiopure pyridino- and piperidino-18-crown-6 ethers. Arkivoc 2016, IV, 130–151. [Google Scholar] [CrossRef] [Green Version]
- Kragl, U.; Dreisbach, C. Continuous Asymmetric Synthesis in a Membrane Reactor. Angew. Chem. Int. Ed. 1996, 35, 642–644. [Google Scholar] [CrossRef]
- Giffels, G.; Beliczey, J.; Felder, M.; Kragl, U. Polymer Enlarged Oxazaborolidines in a Membrane Reactor: Enhancing Effectivity by Retention of the Homogeneous Catalyst. Tetrahedron Asymmetry 1998, 9, 691–696. [Google Scholar] [CrossRef]
- Rissom, S.; Beliczey, J.; Giffels, G.; Kragl, U.; Wandrey, C. Asymmetric Reduction of Acetophenone in Membrane Reactors: Comparison of Oxazaborolidine and Alcohol Dehydrogenase Catalysed Processes. Tetrahedron Asymmetry 1999, 10, 923–928. [Google Scholar] [CrossRef]
- Wöltinger, J.; Bommarius, A.S.; Drauz, K.; Wandrey, C. The Chemzyme Membrane Reactor in the Fine Chemicals Industry. Org. Process Res. Dev. 2001, 5, 241–248. [Google Scholar] [CrossRef]
- Tsogoeva, S.B.; Wöltinger, J.; Jost, C.; Reichert, D.; Kühnle, A.; Krimmer, H.P.; Drauz, K. Juliá-Colonna Asymmetric Epoxidation in a Continuously Operated Chemzyme Membrane Reactor. Synlett 2002, 2002, 707–710. [Google Scholar] [CrossRef]
- Wöltinger, J.; Krimmer, H.P.; Drauz, K. The Potential of Membrane Reactors in the Asymmetric Opening of Meso-Anhydrides. Tetrahedron Lett. 2002, 43, 8531–8533. [Google Scholar] [CrossRef]
- Chavan, S.A.; Maes, W.; Gevers, L.E.M.; Wahlen, J.; Vankelecom, I.F.J.; Jacobs, P.A.; Dehaen, W.; De Vos, D.E. Porphyrin-Functionalized Dendrimers: Synthesis and Application as Recyclable Photocatalysts in a Nanofiltration Membrane Reactor. Chem. Eur. J. 2005, 11, 6754–6762. [Google Scholar] [CrossRef] [PubMed]
- Št’astná, L.C.; Krupková, A.; Petrickovic, R.; Müllerová, M.; Matoušek, J.; Koštejn, M.; Cuřínová, P.; Jandová, V.; Šabata, S.; Strašák, T. Multivalent Bifunctional Carbosilane Dendrimer-Supported Ammonium and Phosphonium Organocatalysts for the Coupling of CO2 and Epoxides. ACS Sustain. Chem. Eng. 2020, 8, 11692–11703. [Google Scholar] [CrossRef]
- Siew, W.E.; Ates, C.; Merschaert, A.; Livingston, A.G. Efficient and Productive Asymmetric Michael Addition: Development of a Highly Enantioselective Quinidine-Based Organocatalyst for Homogeneous Recycling via Nanofiltration. Green Chem. 2013, 15, 663–674. [Google Scholar] [CrossRef]
- Fahrenwaldt, T.; Großeheilmann, J.; Erben, F.; Kragl, U. Organic Solvent Nanofiltration as a Tool for Separation of Quinine-Based Organocatalysts. Org. Process Res. Dev. 2013, 17, 1131–1136. [Google Scholar] [CrossRef]
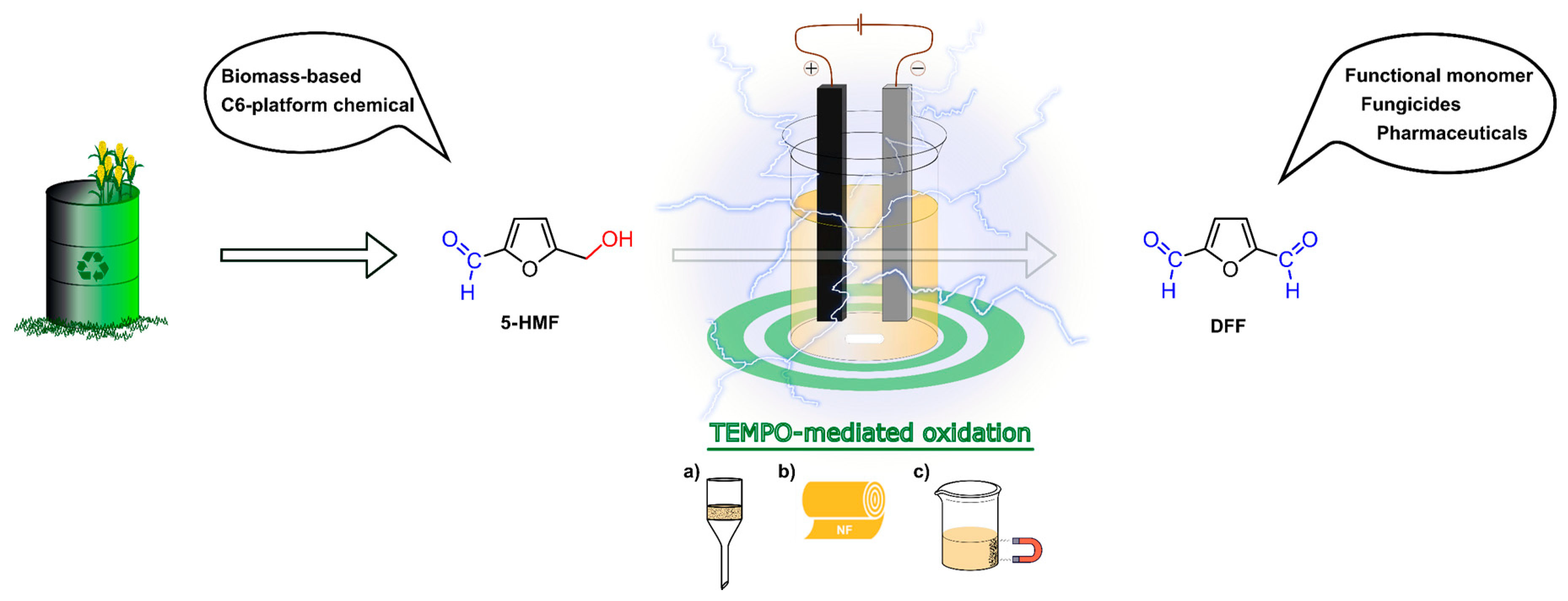
- Kisszekelyi, P.; Hardian, R.; Vovusha, H.; Chen, B.; Zeng, X.; Schwingenschlogl, U.; Kupai, J.; Szekely, G. Selective Electrocatalytic Oxidation of Biomass-derived 5-Hydroxymethylfurfural to 2,5-Diformylfuran: From Mechanistic Investigations to Catalyst Recovery. ChemSusChem 2020, 13, 1–11. [Google Scholar] [CrossRef]
- Kisszekelyi, P.; Alammar, A.; Kupai, J.; Huszthy, P.; Barabas, J.; Holtzl, T.; Szente, L.; Bawn, C.; Adams, R.; Szekely, G. Asymmetric synthesis with cinchona-decorated cyclodextrin in a continuous-flow membrane reactor. J. Catal. 2019, 371, 255–261. [Google Scholar] [CrossRef]
- Gupta, A. Membrane Mediated Organocatalyst Separation Methodology. J. Appl. Chem. 2016, 5, 255–265. [Google Scholar]
- Nagy, S.; Fehér, Z.; Kisszékelyi, P.; Huszthy, P.; Kupai, J. Cinchona derivatives as sustainable and recyclable homogeneous organocatalysts for aza-Markovnikov addition. New J. Chem. 2018, 42, 8596–8602. [Google Scholar] [CrossRef] [Green Version]
- Großeheilmann, J.; Fahrenwaldt, T.; Kragl, U. Organic Solvent Nanofiltration-Supported Purification of Organocatalysts. ChemCatChem 2016, 8, 322–325. [Google Scholar] [CrossRef]
- Großeheilmann, J.; Büttner, H.; Kohrt, C.; Kragl, U.; Werner, T. Recycling of Phosphorus-Based Organocatalysts by Organic Solvent Nanofiltration. ACS Sustain. Chem. Eng. 2015, 3, 2817–2822. [Google Scholar] [CrossRef]
- Min, C.; Han, X.; Liao, Z.; Wu, X.; Zhou, H.-B.; Dong, C. C3-Symmetrical Cinchonine-Squaramide as New Highly Efficient, and Recyclable Organocatalyst for Enantioselective Michael Addition. Adv. Synth. Catal. 2011, 353, 2715–2720. [Google Scholar] [CrossRef]
- Skowroński, R.; Cottier, L.; Descotes, G.; Lewkowski, J. Selective Anodic Oxidation of 5-Hydroxymethylfurfural. Synthesis 1996, 1996, 1291–1292. [Google Scholar] [CrossRef]
- Cao, T.; Wu, M.; Ordomsky, V.V.; Xin, X.; Wang, H.; Métivier, P.; Pera-Titus, M. Selective Electrogenerative Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandialdehyde. ChemSusChem 2017, 10, 4851–4854. [Google Scholar] [CrossRef]
- Delorme, A.E.; Sans, V.; Licence, P.; Walsh, D.A. Tuning the Reactivity of TEMPO during Electrocatalytic Alcohol Oxidations in Room-Temperature Ionic Liquids. ACS Sustain. Chem. Eng. 2019, 7, 11691–11699. [Google Scholar] [CrossRef]
- Rafiee, M.; Konz, Z.M.; Graaf, M.D.; Koolman, H.F.; Stahl, S.S. Electrochemical Oxidation of Alcohols and Aldehydes to Carboxylic Acids Catalyzed by 4-Acetamido-TEMPO: An Alternative to “Anelli” and “Pinnick” Oxidations. ACS Catal. 2018, 8, 6738–6744. [Google Scholar] [CrossRef]
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
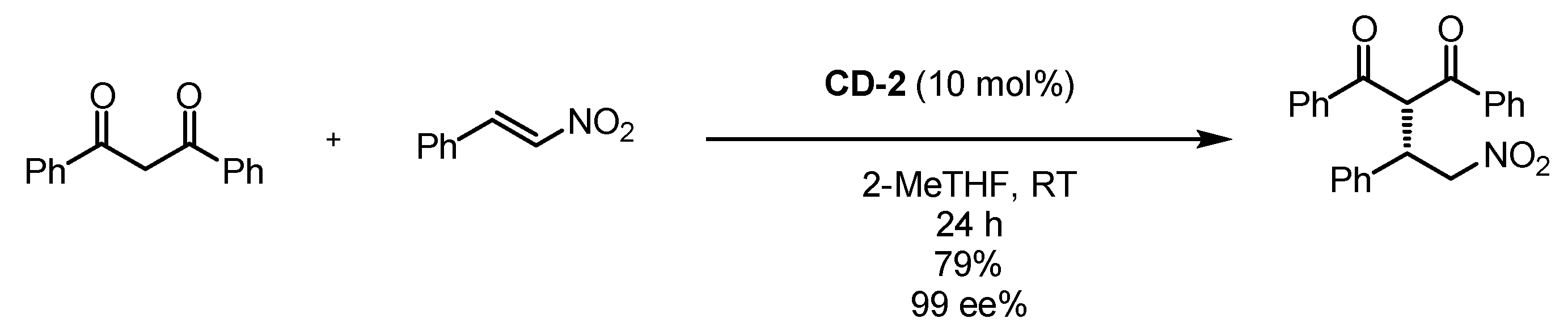{kind=link}
| Process Type | Pore Size of Membrane (nm) | Examples for Application |
|---|---|---|
| microfiltration | 50–500 | yeast, fungus, bacteria, oil emulsion |
| ultrafiltration | 2–50 | colloidal solid, virus, protein, polysaccharide |
| nanofiltration | ≤2 | catalysts, dyes, antibiotics, API impurities |
| reverse osmosis | 0.3–0.6 | water, inorganic ions |
| Reference | Type of Molecular Weight Enlargement | Catalyst MW [g mol−1] | Membrane Type a | Solvent b | Catalyst Retention [%] |
|---|---|---|---|---|---|
| Kupai et al. [46] | - | 351–435 | PBI | toluene | 97–100 |
| Kisszekelyi et al. [45] | - | 322–538 | PBI | IPA, THF, toluene | 48–99 |
| Nagy et al. [60] | - | 325 | GMT-oNF-1, -2, -3, PBI | MeCN | 88–99 |
| Großeheilmann et al. [62] | - | 374 | DM150, 200, 300, 500 | EtOH, acetone, butylene carbonate | 84–99 |
| Kragl et al. [47] | soluble polymer | ~96,000 | Nadir UF PA20 | n-hexane | 100 |
| Giffels et al. [48] | soluble polymer | ~13,800 | MPF-50 | THF | n.a. |
| Rissom et al. [49] | soluble polymer | ~14,000 | MPF-50 | THF | n.a. |
| Wöltinger et al. [50] | soluble polymer | ~22,640 | MPF-50 | THF | 99–100 |
| Tsogoeva et al. [51] | soluble polymer | n.a. | MPF-50 | THF | 99 |
| Wöltinger et al. [52] | soluble polymer | n.a. | n.a. | toluene:MeOH | n.a. |
| Chavan et al. [53] | dendrimer | 2117–8650 | MPF-50, PDMS, PDMS-USY-PAN | IPA, CHCl3 | 40–99 |
| Št’astná et al. [54] | dendrimer | 1342–7724 | regenerated cellulose | MeOH | 20–98 c |
| Siew et al. [55] | multifunctional core | 1044–1332 | DM500, 300 | THF | >99 |
| Kisszekelyi et al. [57] | multifunctional core | 631 | GMT-oNF-1, NF030306, DM300 | MeCN | 90–100 |
| Fahrendwaldt et al. [56] | benzoyl subunit | 324–429 | DM150, 200, 300, 500 | THF | 90–100 |
| Kisszekelyi et al. [58] | cyclodextrin-anchoring | 1817 | GMT-oNF-1, NF030306, NF010306, DM300, 500, 900 | 2-MeTHF | 98–100 |
| Großeheilmann et al. [61] | esterification | 409–521 | DM200, 300 | EtOH | 53–100 |
| Gupta [59] | salt formation | 115–548 | PDCPD | DCM, MeOH | 0–99 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kisszékelyi, P.; Nagy, S.; Fehér, Z.; Huszthy, P.; Kupai, J. Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review. Chemistry 2020, 2, 742-758. https://doi.org/10.3390/chemistry2030048
Kisszékelyi P, Nagy S, Fehér Z, Huszthy P, Kupai J. Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review. Chemistry. 2020; 2(3):742-758. https://doi.org/10.3390/chemistry2030048
Chicago/Turabian StyleKisszékelyi, Péter, Sándor Nagy, Zsuzsanna Fehér, Péter Huszthy, and József Kupai. 2020. "Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review" Chemistry 2, no. 3: 742-758. https://doi.org/10.3390/chemistry2030048
APA StyleKisszékelyi, P., Nagy, S., Fehér, Z., Huszthy, P., & Kupai, J. (2020). Membrane-Supported Recovery of Homogeneous Organocatalysts: A Review. Chemistry, 2(3), 742-758. https://doi.org/10.3390/chemistry2030048