Stereosopecificity in [Co(sep)][Co(edta)]Cl2·2H2O


Abstract
:1. Introduction
2. Materials and Methods
3. Results
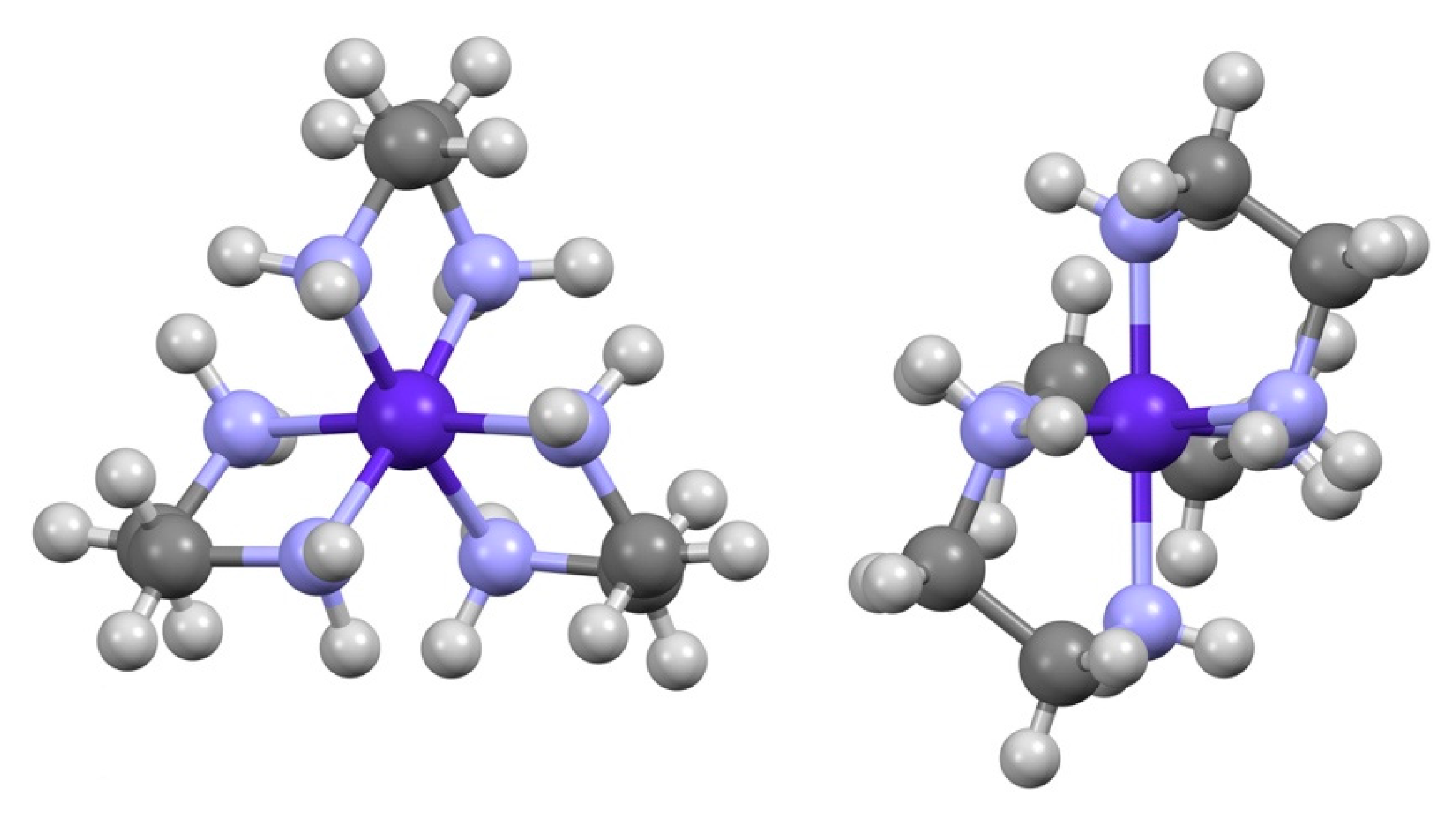
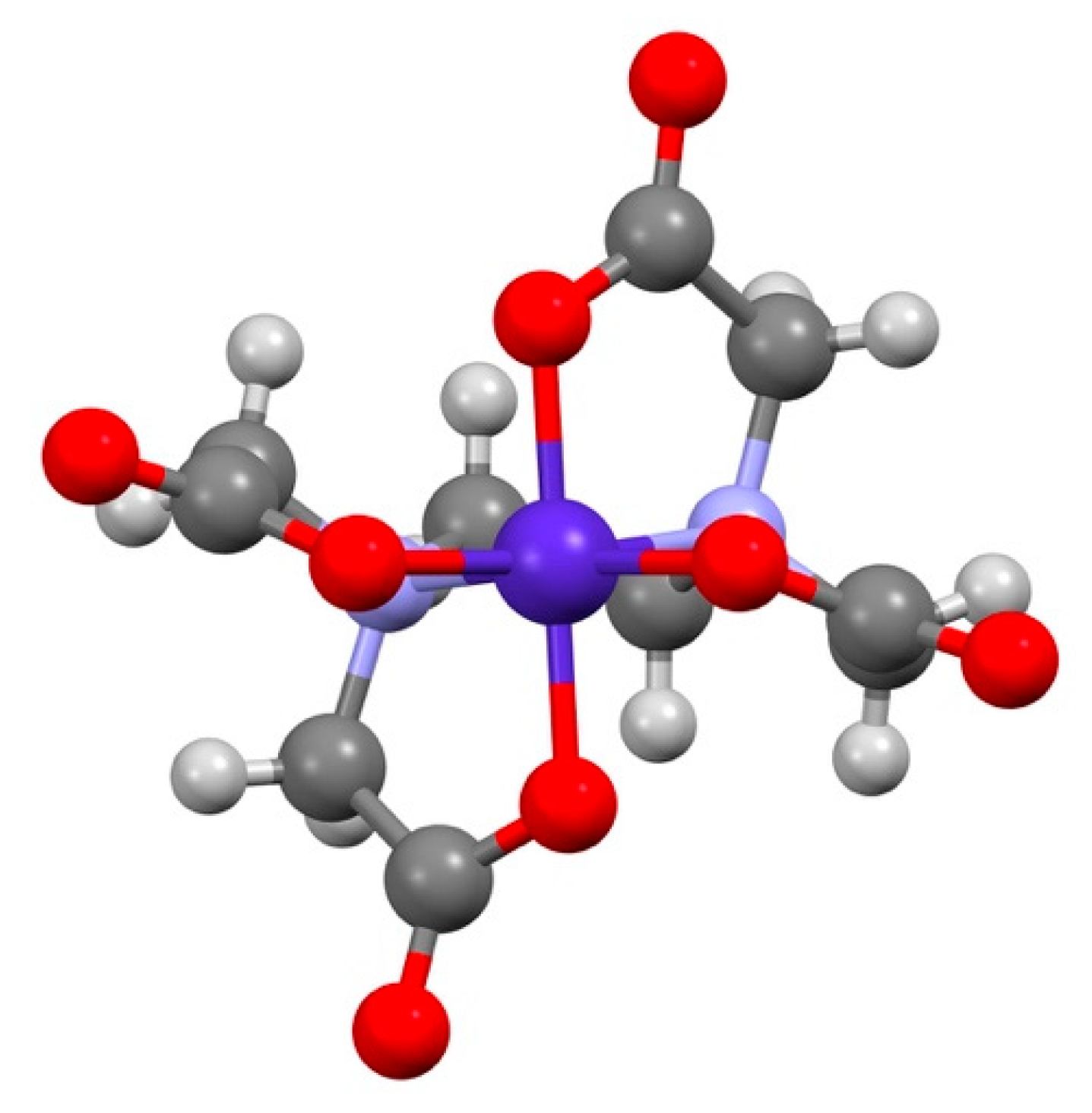
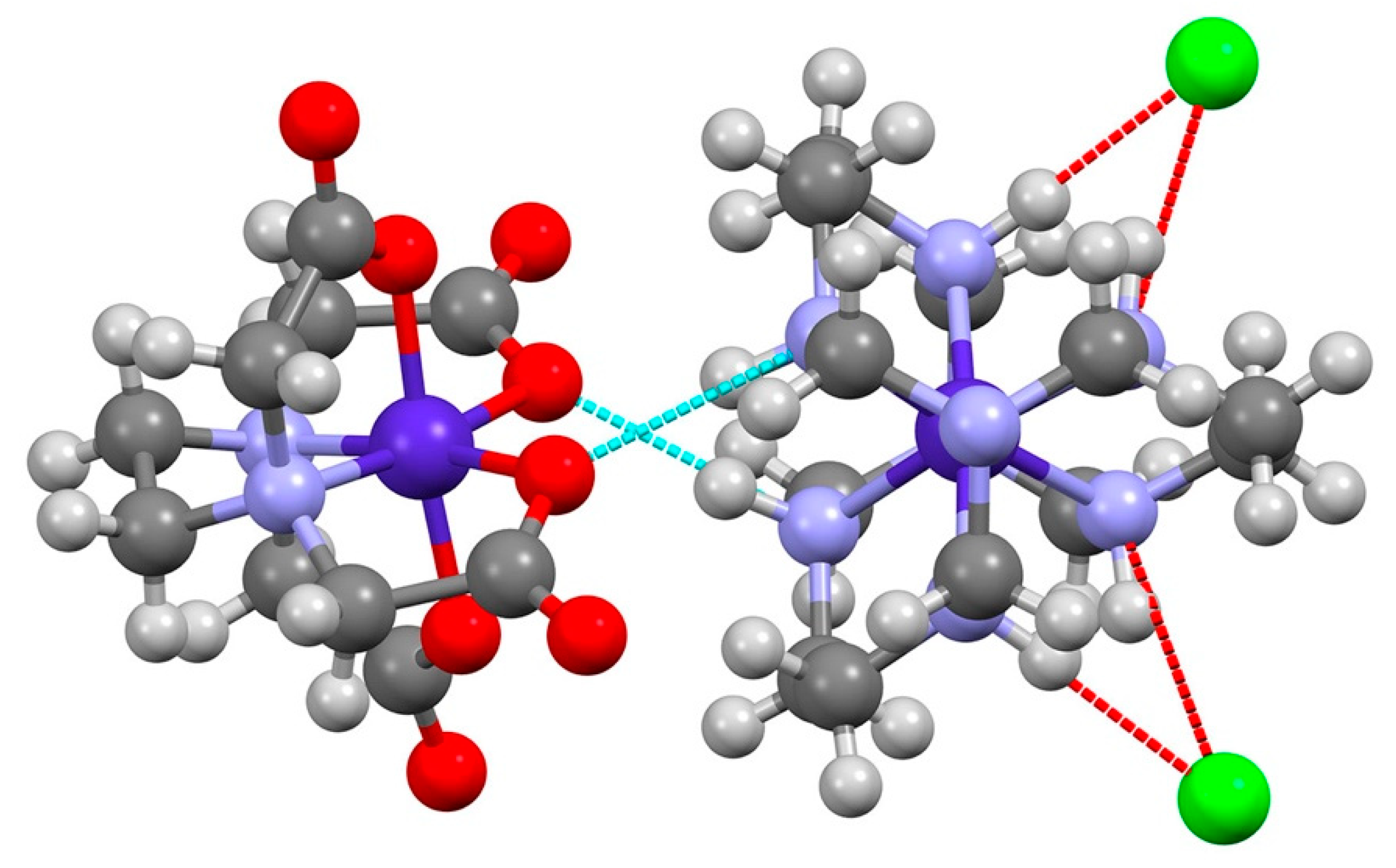
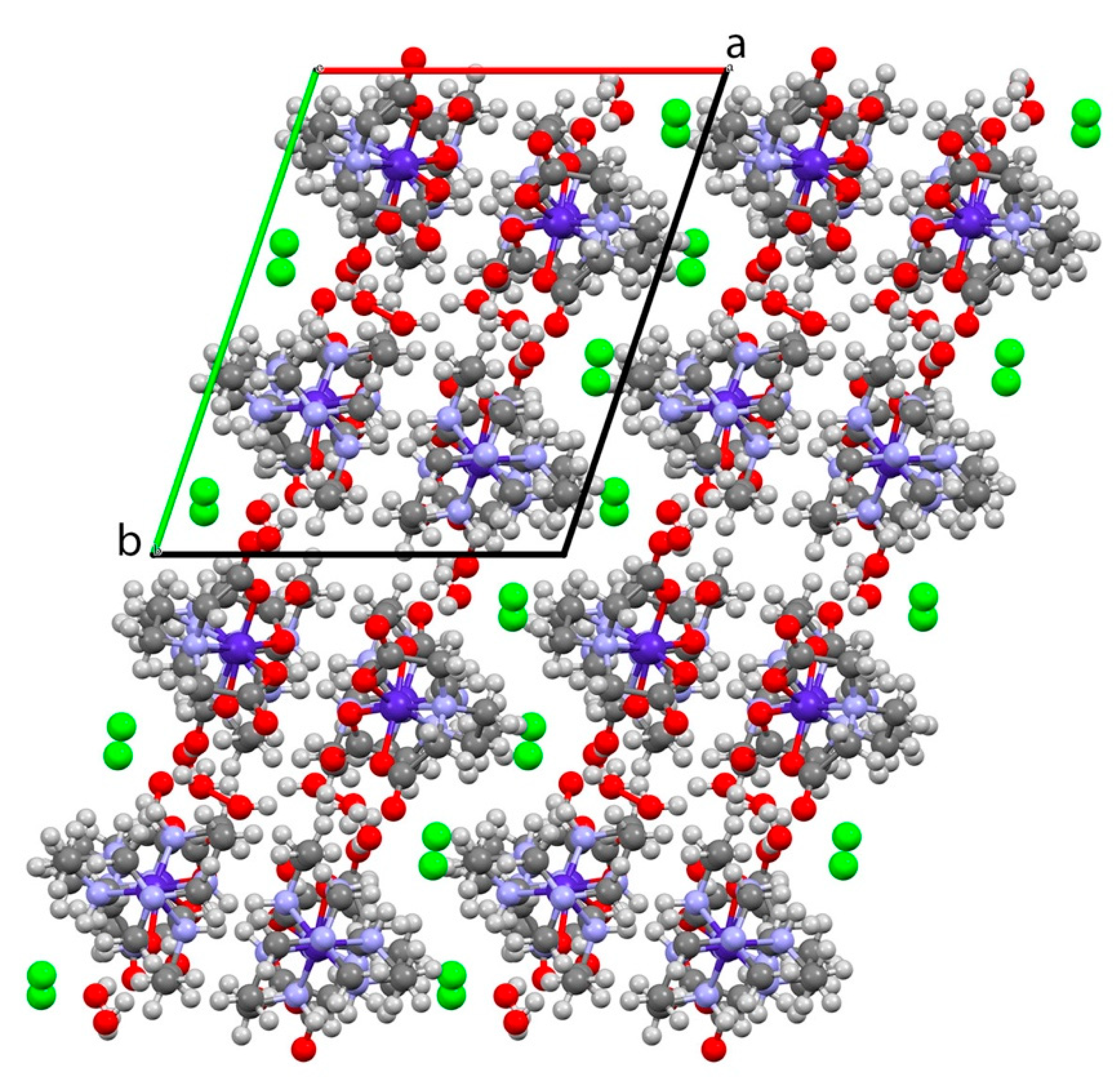
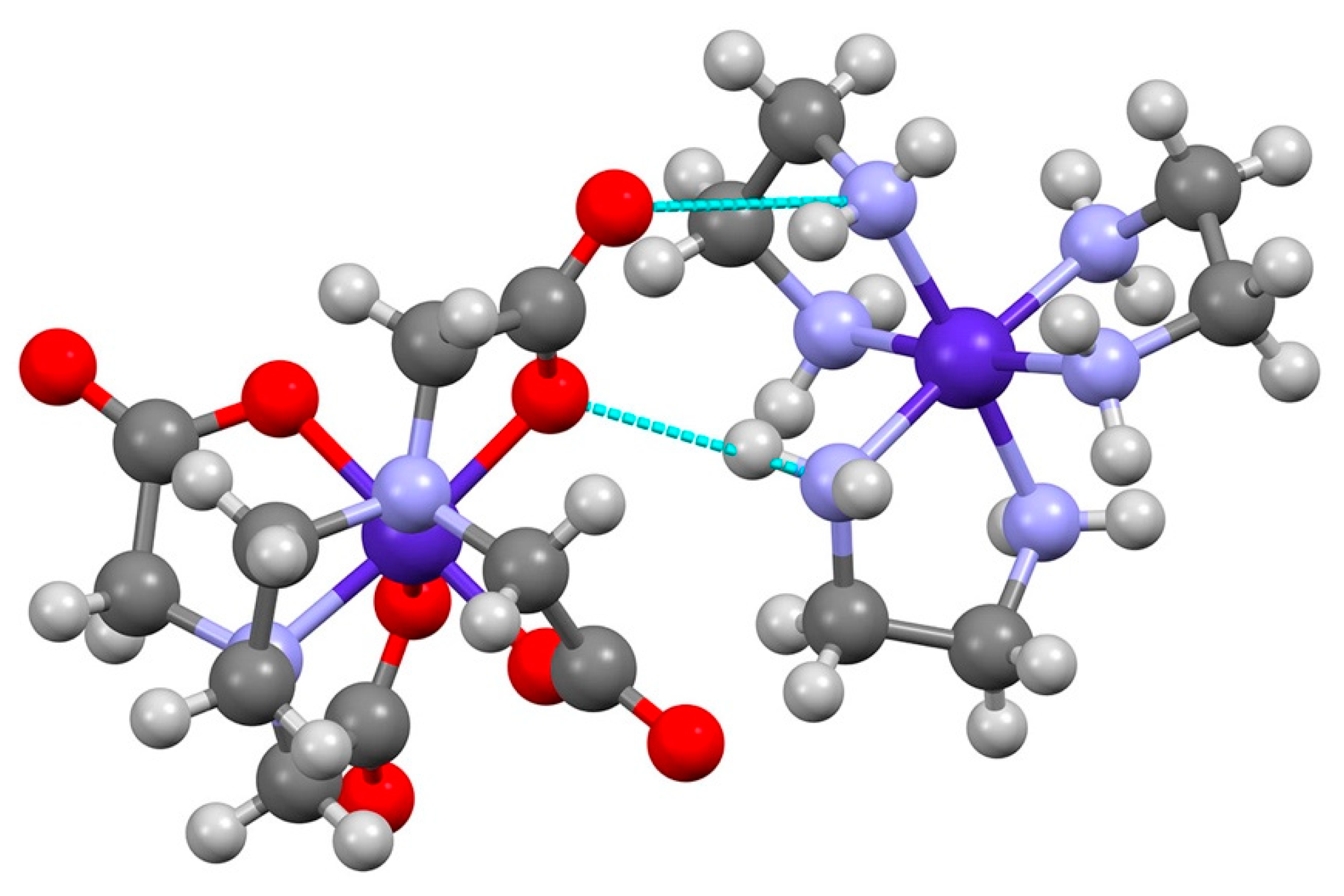
3.1. [Co(sep)][Co(edta)]Cl2·2H2O
3.2. (Λ-[Co(en)3]∆-[Co(edta)]2Cl·10(H2O)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flack, H.D. On Enantiomorph-Polarity Estimation. Acta Crystallogr. 1983, A39, 876–881. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Yamamoto, I.; Izumoto, S.; Yoneda, H. The Mode of Stereoselective Association between Complex Cation and Complex Anion. Bull. Chem. Soc. Jpn. 1983, 56, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, K.; Sakamoto, Y.; Ohguni, A.; Yoneda, H. Stereoselective Interaction of Chiral Metal Complexes in Solution as Studied by Chromatography. i. Modes of Chiral Discrimination and Optical Resolution of anion Complexes. Bull. Chem. Soc. Jpn. 1985, 58, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Tatehata, A.; Fujita, M.; Ando, K.; Asaba, Y. Chiral Recognition in Ion Pairing of an Optically Active Tris(Ethylenediamine)Cobalt(Iii) Cation and Applications to Chromatographic Resolution of Metal Complex Anions. J. Chem Soc. Dalton Trans. 1987, 8, 1977–1982. [Google Scholar] [CrossRef]
- Marusak, R.A.; Lappin, A.G. Observations on the Structure of the Ion Pair Formed between [Co(en)3]3+ and [Co(edta)]–. J. Phys. Chem. 1989, 93, 6856–6859. [Google Scholar] [CrossRef]
- Dwyer, F.P.; Gyarfas, E.C.; Mellor, D.P. The Resolution and Racemization of Potassium Ethylenediaminetetraacetatocobaltate (III). J. Phys Chem. 1955, 59, 296–297. [Google Scholar] [CrossRef]
- Geselowitz, D.A.; Taube, H. Stereoselectivity in Electron-Transfer Reactions. J. Am. Chem Soc. 1980, 102, 4525–4526. [Google Scholar] [CrossRef]
- Osvath, P.; Lappin, A.G. Conformational Effects in Stereoselective Electron Transfer between Metal Ion Complexes. J. Chem. Soc. Chem. Commun. 1986, 14, 1056–1057. [Google Scholar] [CrossRef]
- Osvath, P.; Lappin, A.G. Stereoselectivity as a Probe of Reaction Mechanism in the Oxidation of [Co(en)3]2+ and Its Derivatives by [Co(edta)]–. Inorg. Chem. 1987, 26, 195–202. [Google Scholar] [CrossRef]
- Geselowitz, D.A.; Hammershøi, A.; Taube, H. Stereoselective Electron-Transfer Reactions of (Ethylenediaminetetraacetato)cobaltate(III), (Propylenediaminetetraacetato)cobaltate(III), and (1,2-Cyclohexanediaminetetraacetato)cobaltate(III) with Tris(ethylenediamine)cobalt(II). Inorg. Chem. 1987, 26, 1842–1845. [Google Scholar] [CrossRef]
- Tatehata, A.; Mitani, T. Stereoselectivity in Electron-Transfer Reactions of Tris(ethylenediamine)cobalt(II) with Several Anionic Cobalt(III) Complexes. Chem. Lett. 1989, 18, 1167–1170. [Google Scholar] [CrossRef]
- Tatehata, A.; Muraida, A. Temperature Dependence of Chiral Recognition in the Oxidation of Tris(ethylenediamine)cobalt(II) Complex by Optically Active Anionic Cobalt(III) Complexes. Chem. Lett. 1996, 25, 461–462. [Google Scholar] [CrossRef]
- Warren, R.M.L.; Haller, K.J.; Tatehata, A.; Lappin, A.G. Chiral Discrimination in the Reduction of [Co(edta)]– by [Co(en)3]2+ and [Ru(en)3]2+. X-ray Structure of [Λ-Co(en)3][Δ-Co(edta)]2Cl.10H2O. Inorg. Chem. 1994, 33, 227–232. [Google Scholar] [CrossRef]
- Harrowfield, J.M.; Herlt, A.J.; Sargeson, A.M. Caged Metal Ions: Cobalt Sepulchrates. Inorg. Synth. 1980, 20, 85–86. [Google Scholar]
- APEX-3; Bruker AXS: Madison, WI, USA, 2016.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Crystal data for C22H46Cl2Co2N10O10; Mr = 799.45; Triclinic; space group P-1; a = 13.0187(8) Å; b = 15.4433(10) Å; c = 17.2326(11) Å; α = 90.310(2)°; β = 108.030(2)°; γ = 107.509(2)°; V = 3123.3(3) Å3; Z = 4; T = 150(2) K; λ(synchrotron) = 0.72880 Å; μ(synchrotron) = 1.387 mm−1; dcalc = 1.700 g.cm−3; 162884 reflections collected; 13890 unique (Rint = 0.1250); giving R1 = 0.0561, wR2 = 0.1480 for 10514 data with [I > 2σ(I)] and R1 = 0.0768, wR2 = 0.1643 for all 13890 data. Residual electron density (e–·Å−3) max/min: 2.239/-0.632. Available online: https://rruff.geo.arizona.edu/AMS/amcsd.php (accessed on 6 February 2021).
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Cryst. 2013, B69, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Hooft, R.W.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Cryst. 2008, 41, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Crystal data for C26H68ClCo3N10O26; Mr = 1149.14; Orthorhombic; space group P21212; a = 12.8265(17) Å; b = 21.055(3) Å; c = 8.1536(11) Å; α = 90°; β = 90°; γ = 90°; V = 2202.0(5) Å3; Z = 2; T = 120(2) K; λ(Mo-Kα) = 0.71073 Å; μ(Mo-Kα) = 1.280 mm−1; dcalc = 1.733 g.cm−3; 42751 reflections collected; 5478 unique (Rint = 0.0550); giving R1 = 0.0271, wR2 = 0.0642 for 5032 data with [I > 2σ(I)] and R1 = 0.0323, wR2 = 0.0662 for all 5478 data. Residual electron density (e–·Å−3) max/min: 0.500/-0.533. Available online: https://rruff.geo.arizona.edu/AMS/amcsd.php (accessed on 6 February 2021).
- Sysoeva, T.F.; Agre, V.M.; Trunov, V.K.; Dyatlova, N.M.; Fridman, A.Y. Crystal structure of complex compound of nickel(II) with ethylenediamine and ethylenediaminetetraacetic acid, Ni2en3A.4H2O. J. Struct. Chem. (Engl. Transl.) 1986, 27, 97–103. [Google Scholar] [CrossRef]
- Tembe, B.L.; Friedman, H.L.; Newton, M.D. The Theory of Fe2+–Fe3+ Electron Exchange in Water. J. Chem. Phys. 1982, 76, 1490–4367. [Google Scholar] [CrossRef]
- Logan, J.; Newton, M.D. Ab initio study of electronic coupling in the aqueous Fe2+-Fe3+ electron exchange process. J. Chem. Phys. 1983, 78, 4086–4091. [Google Scholar] [CrossRef]
- Newton, M.D. Electronic Structure Analysis of Electron-Transfer Matrix Elements for Transition-Metal Redox Pairs. J. Phys. Chem. 1988, 92, 3049–3056. [Google Scholar] [CrossRef]
- Kumar, P.V.; Tembe, B.L. Solvation structure and dynamics of the Fe2+–Fe3+ ion pair in water. J. Chem. Phys. 1992, 97, 4356–4367. [Google Scholar] [CrossRef]
- Babu, C.S.; Madhusoodanan, M.; Sridhar, G.; Tembe, B.L. Orientations of [Fe(H2O)6]2+ and [Fe(H2O)6]3+ Complexes at a Reactive Separation in Water. J. Am. Chem. Soc. 1997, 119, 5679–5681. [Google Scholar] [CrossRef]
- Migliore, A.; Sit, P.H.-L.; Klein, M.L. Evaluation of Electronic Coupling in Transition-Metal Systems Using DFT: Application to the Hexa-Aquo Ferric-Ferrous Redox Couple. J. Chem. Theory Comput. 2009, 5, 307–323. [Google Scholar] [CrossRef]
- Oberhofer, H.; Blumberger, J. Insight into the Mechanism of the Ru2+–Ru3+ Electron Self-Exchange Reaction from Quantitative Rate Calculations. Angew. Chem. Int. Ed. 2010, 49, 3631–3634. [Google Scholar] [CrossRef]
- Miliordos, E.; Xantheas, S.S. Ground and Excited States of the [Fe(H2O)6]2+ and [Fe(H2O)6]3+ Clusters: Insight into the Electronic Structure of the [Fe(H2O)6]2+-[Fe(H2O)6]3+ Complex. J. Chem. Theory Comput. 2015, 11, 1549–1563. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Larsson, K.; Neal, T.J.; Wyllie, G.R.A.; Shang, M.; Lappin, A.G. Structure and magnetic properties of [Cr(en)2(ox)][Cr(en)(ox)2]·2H2O, Δ-[Cr(en)2(ox)]Δ-[Cr(en)(ox)2] and Δ-[Cr(en)2(ox)]Δ-[Cr(en)(ox)2]. Inorg. Chem. Commun. 2001, 4, 635–639. [Google Scholar] [CrossRef]
- Mitani, T.; Honma, N.; Tatehata, A.; Lappin, A.G. Shape selectivity in outer-sphere electron transfer reactions. Inorg. Chim. Acta 2002, 331, 39–71. [Google Scholar] [CrossRef]
- Saenz, G.L.; Warren, R.M.L.; Shang, M.; Lappin, A.G. Stereoselectivity in the Reduction of Λ-[1,4,7-Triazacyclononane-1,4,7-tris[2’(R)-2’-propionate]cobalt(III)]. J. Coord Chem. 1995, 34, 129–137. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D-H···A | d(D-H) | d(H···A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| Λ-[Co(sep)]∆-[Co(edta)]Cl2 | ||||
| N(7)-H(7)···Cl(1) | 1.00 | 2.21 | 3.204(3) | 175.2 |
| N(3)-H(3)···Cl(1) | 1.00 | 2.27 | 3.215(3) | 157.5 |
| N(2)-H(2)···Cl(2) | 1.00 | 2.19 | 3.138(3) | 158.0 |
| N(6)-H(6)···Cl(2) | 1.00 | 2.17 | 3.171(3) | 174.9 |
| N(5)-H(5)···O(1) | 1.00 | 2.03 | 2.919(4) | 146.8 |
| N(8)-H(8)···O(5) | 1.00 | 2.03 | 2.947(4) | 151.7 |
| ∆-[Co(sep)]Λ-[Co(edta)]Cl2 | ||||
| N(10)-H(10)···Cl(3) | 1.00 | 2.11 | 3.075(3) | 162.9 |
| N(16)-H(16)···Cl(3) | 1.00 | 2.36 | 3.346(3) | 168.1 |
| N(11)-H(11)···Cl(4) | 1.00 | 2.17 | 3.139(3) | 163.6 |
| N(13)-H(13)···Cl(4) | 1.00 | 2.25 | 3.238(3) | 169.4 |
| N(14)-H(14)···O(15) | 1.00 | 2.02 | 2.908(4) | 147.4 |
| N(15)-H(15)···O(9) | 1.00 | 1.97 | 2.883(4) | 151.0 |
| Λ-[Co(en)3]∆-[Co(edta)]2Cl | ||||
| G-ring interactions | ||||
| N(1)-H(1NA)···O(5)#2 a | 0.87(4) | 3.07(4) | 3.757(4) | 137(3) |
| N(1)-H(1NA)···O(6)#2 b | 0.87(4) | 2.23(4) | 3.009(3) | 149(3) |
| N(3)-H(3NB)···O(5)#4(ob) a | 0.91 | 2.10 | 2.985(3) | 162.8 |
| N(3)-H(3NB)···O(6)#4(ob) b | 0.91 | 3.13 | 3.783(3) | 129.9 |
| N(1)-H(1NA)···O(5)#2 a | 0.87(4) | 3.07(4) | 3.757(4) | 137(3) |
| N(1)-H(1NA)···O(6)#2 b | 0.87(4) | 2.23(4) | 3.009(3) | 149(3) |
| R-ring interactions | ||||
| N(1)-H(1NA)···O(2)#1 b | 0.87(4) | 3.02(4) | 3.623(4) | 129(3) |
| N(1)-H(1NB)···O(8)#3 b | 0.77(5) | 3.08(5) | 3.558(4) | 123(4) |
| N(3)-H(3NC)···O(2)(lel) b | 0.91 | 1.96 | 2.850(3) | 165.1 |
| N(3)-H(3NA)···O(8)#4(ob) b | 0.91 | 3.39 | 3.820(3) | 111.4 |
| N(3)-H(3ND)···O(7)#4(lel) a | 0.91 | 3.02 | 3.489(3) | 114.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osvath, P.; Oliver, A.; Lappin, A.G. Stereosopecificity in [Co(sep)][Co(edta)]Cl2·2H2O. Chemistry 2021, 3, 228-237. https://doi.org/10.3390/chemistry3010017
Osvath P, Oliver A, Lappin AG. Stereosopecificity in [Co(sep)][Co(edta)]Cl2·2H2O. Chemistry. 2021; 3(1):228-237. https://doi.org/10.3390/chemistry3010017
Chicago/Turabian StyleOsvath, Peter, Allen Oliver, and A. Graham Lappin. 2021. "Stereosopecificity in [Co(sep)][Co(edta)]Cl2·2H2O" Chemistry 3, no. 1: 228-237. https://doi.org/10.3390/chemistry3010017
APA StyleOsvath, P., Oliver, A., & Lappin, A. G. (2021). Stereosopecificity in [Co(sep)][Co(edta)]Cl2·2H2O. Chemistry, 3(1), 228-237. https://doi.org/10.3390/chemistry3010017