All chemicals were purchased from Millipore Sigma and used without further purification. All synthesized compounds were purified using flash column chromatography. 1H-NMR and 13C-NMR were recorded at 300 MHz on a Varian instrument using VnmrJ version 4.2A. NMR spectroscopy for compounds 21–24 was performed on a Bruker AVANCE III 400 MHz spectrometer. For in vitro studies, a fluorometric assay was performed in 96 non-binding microplates from Greiner Bio-One with a clear bottom on a Synergy Bio-tek HTS plate reader.
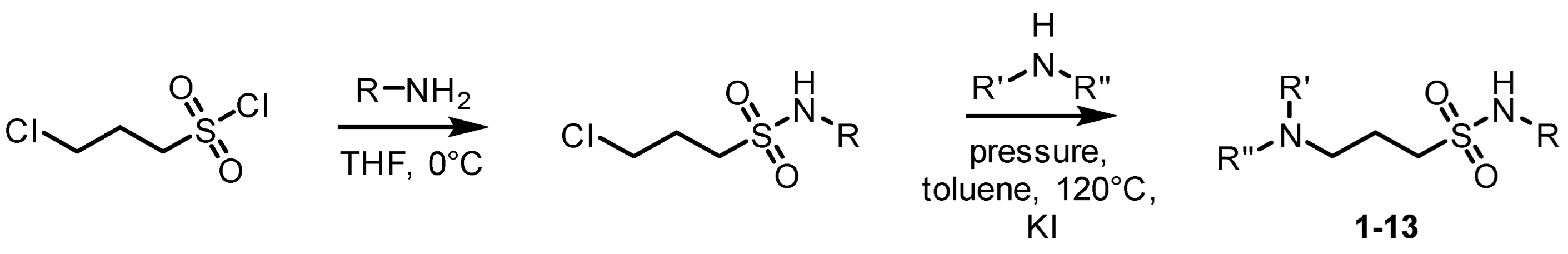
4.1. Preparation and Characterization of Homotaurine and Curcumin Analogues
3-chloropropanesulfonyl chloride (0.89 g, 5.03 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby isobutyl amine (4 mL, 0.04 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and isobutyl amine (0.89 mL, 8.95 mmol) were added and the mixture was heated to 130 °C for 48 h. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.52 g, 42%). 1H NMR (300 MHz, CDCl3) δ 0.90 (d, J = 6.7 Hz, 6H), 0.95 (d, J = 6.7 Hz, 6H), 1.76 (m, 2H), 1.98 (quint, J = 6.5 Hz, 1H), 2.40 (d, J = 6.8 Hz, 2H), 2.73 (t, J = 6.4 Hz, 2H), 2.91 (d, J = 6.8 Hz, 2H), 3.11 (t, J = 7.3 Hz, 2H). 13C NMR (75 MHz, DMSO-d) δ 54.91, 50.25, 48.82, 46.53, 28.86, 26.25, 21.18, 20.68, 20.40.
- 2.
N-(isobutyl)-3-(diethylamino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (0.58 g, 3.27 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby isobutyl amine (4 mL, 0.04 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and diethyl amine (0.42 mL, 5.7 mmol) were added and the mixture was heated to 130 °C for 48 h. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.15 g, 18%). 1H-NMR (300 MHz, CDCl3) δ 0.95 (d, J = 6.7 Hz, 6H), 1.00 (t, J = 7.2 Hz, 6H), 1.78 (m, 1H), 1.94 (quint, J = 7.4 Hz, 2H), δ 2.52 (m, 6H), δ 2.90 (t, J = 5.2 Hz, 2H), 3.07 (t, J = 7.6 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 51.14, 50.72, 50.54, 46.48, 29.00, 21.54, 19.89, 11.42.
- 3.
N-(t-butyl)-3-(diethylamino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (1.52 g, 8.57 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby t-butyl amine (4 mL, 0.04 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and diethyl amine (2 mL, 19.2 mmol) were added and the mixture was heated to 130 °C for 48 h. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (1.00 g, 46%). 1H-NMR (300 MHz, CDCl3) δ 0.98 (t, J = 7.15 Hz, 6H), 1.36 (s, 9H), 1.92 (quint, J = 7.2 Hz, 2H), 2.5 (m, 6H), 3.06 (t, J = 7.7 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 54.47, 54.40, 51.05, 46.56, 30.34, 22.00, 11.61.
- 4.
N-butyl-3-(diethylamino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (1.46 g, 8.22 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby n-butyl amine (4 mL, 0.04 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and diethyl amine (0.42 mL, 5.7 mmol) were added and the mixture was heated to 130 °C for 48 hrs. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (1.46 g, 71%). 1H-NMR (300 MHz, CDCl3) δ 0.91 (t, J = 7.4 Hz, 3H), 0.98 (t, J = 7.1 Hz 6H), 1.36 (m, 2H), 1.52 (quint, J = 7.8 Hz, 2H), δ 1.92 (quint, J = 7.2 Hz, 2H), 2.50 (m, 6H), 3.06 (m, 4H). 13C-NMR (75 MHz, CDCl3) δ 51.23, 50.84, 46.46, 42.93, 32.41, 21.63, 19.78, 13.63, 11.46.
- 5.
N-isopropyl-3-(diethylamino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (0.59 g, 3.33 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby isopropyl amine (4 mL, 0.05 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and diethyl amine (1 mL, 9.6 mmol) were added and the mixture was heated to 130 °C for 48 h. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.55 g, 70%). 1H-NMR (300 MHz, CDCl3) δ 1.02 (d, J = 7.4 Hz, 6H), δ 2.04 (quint, J = 7.2 Hz, 2H), 2.93 (t, J = 7.6 Hz, 2H), 3.35 (m, 1H), 3.5 (m, 4H).
- 6.
N-isobutyl-3-(dipropylamino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (1.30 g, 7.32 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby isobutyl amine (4 mL, 0.04 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and dipropyl amine (2 mL, 14.62 mmol) were added and the mixture was heated to 130 °C for 48 h. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.41 g, 20%). 1H-NMR (300 MHz, CDCl3) δ 0.86 (t, J = 7.3 Hz, 6H), 0.95 (d, J = 6.7 Hz, 6H), 1.42 (sextet, J = 7.4 Hz, 4H), 1.79 (m, 1H), 1.92 (m, 2H), 2.35 (t, J = 7.4 Hz, 4H), 2.51 (t, J = 6.5 Hz, 2H), 2.92 (t, J = 6.3 Hz, 2H), 3.08 (t, J = 7.6 Hz, 2H). Yield: 70%. 13C-NMR (75 MHz, CDCl3) δ 55.83, 52.41, 50.84, 50.61, 29.01, 21.84, 20.10, 19.88, 11.91.
- 7.
N-isopropyl-3-(dipropylamino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (1.01 g, 5.70 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby isopropyl amine (4 mL, 0.05 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and dipropyl amine (2 mL, 14.62 mmol) were added and the mixture was heated to 130 °C for 48 hrs. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.22 g, 14%). 1H-NMR (300 MHz, CDCl3) δ 0.82 (t, J = 7.4 Hz, 6H), 1.19 (d, J = 6.5 Hz, 6H), 1.39 (m, 4H), 1.88 (quint, J = 6.8 Hz, 2H), 2.32 (t, J = 7.5 Hz, 4H), 2.47 (t, J = 6.4 Hz, 2H), 3.04 (t, J = 7.8 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 55.68, 52.16, 51.94, 46.00, 24.19, 21.73, 19.99, 11.83, 11.81, 11.79.
- 8.
N-butyl-3-(dipropylamino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (1.31 g, 7.39 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby n-butyl amine (4 mL, 0.04 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and dipropyl amine (2 mL, 14.62 mmol) were added and the mixture was heated to 130 °C for 48 hrs. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.19 g, 10%). 1H-NMR (300 MHz, CDCl3) δ 0.85 (t, J = 7.3 Hz, 6H), 0.91 (t, J = 7.3 Hz, 3H), 1.40 (m, 6H), 1.53 (quint, J = 7.6 Hz, 2H), 1.92 (quint, J = 7.5 Hz, 2H), 2.36 (t, J = 7.5 Hz, 4H), 2.52 (t, J = 6.5 Hz, 2H), 3.07 (m, 4H). 13C-NMR (75 MHz, CDCl3) δ 55.64, 52.27, 50.68, 42.98, 32.39, 21.67, 19.89, 19.79, 13.64, 11.90.
- 9.
N-(t-butyl)-3-(dipropylamino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (1.33 g, 7.52 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby t-butyl amine (4 mL, 0.04 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and dipropyl amine (2 mL, 14.62 mmol) were added and the mixture was heated to 130 °C for 48 h. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.74 g, 35%). 1H-NMR (300 MHz, CDCl3) δ 0.86 (t, J = 7.3 Hz, 6H), 1.41 (m, 13H), 1.92 (quint, J = 6.8 Hz, 2H), 2.35 (t, J = 7.5 Hz, 4H), 2.50 (t, J = 6.8 Hz, 2H), 3.10 (t, J = 7.8 Hz, 2H), 4.32 (s, 1H). 13C-NMR (75 MHz, CDCl3) δ 55.86, 54.44, 54.40, 52.31, 30.33, 22.14, 20.17, 11.91.
- 10.
N-isopropyl-3-(butyl(ethyl)amino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (1.43 g, 8.07 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby isopropyl amine (4 mL, 0.05 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and N-ethylbutyl amine (2 mL, 16.75 mmol) were added and the mixture was heated to 130 °C for 48 h. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate:hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.22 g, 10%). 1H-NMR (300 MHz, CDCl3) δ 0.92 (t, J = 7.5 Hz, 3H), 1.04 (t, J = 7.0 Hz, 3H), 1.24 (d, J = 6.5 Hz, 6H), 1.29 (m, 2H), 1.44 (m, 2H), 1.99 (quint, J = 7.2 Hz, 2H), 2.48 (t, J = 7.8 Hz, 2H), 2.60 (m, 4H), 3.08 (t, J = 7.5 Hz, 2H), δ 3.64 (quint, J = 6.5 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 52.70, 52.03, 51.46, 47.01, 46.18, 28.49, 24.34, 21.46, 20.61, 14.02, 11.09.
- 11.
N-benzyl-3-(t-butylamino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (1.22 g, 6.89 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby benzyl amine (4 mL, 0.03 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and t-butyl amine (2 mL, 19.03 mmol) were added and the mixture was heated to 130 °C for 48 hrs. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate:hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (1.07 g, 54%). 1H-NMR (300 MHz, CDCl3) δ 1.01 (s, 9H), 1.92 (quint, J = 6.8 Hz, 2H), 2.63 (t, J = 6.2 Hz, 2H), 3.07 (t, J = 6.2 Hz, 2H), 3.69 (s, 1H), 4.28 (d, J = 7.8 Hz, 2H), 7.32 (m, 5H).
- 12.
N-isopropyl-3-(butyl(methyl)amino)propane-1-sulfonamide
3-chloropropanesulfonyl chloride (0.88 g, 4.96 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby isopropyl amine (4 mL, 0.05 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and N-methylbutyl amine (1.6 mL, 12.66 mmol) were added and the mixture was heated to 130 °C for 48 h. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate:hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.16 g, 9%). 1H-NMR (300 MHz, CDCl3) δ 0.89 (t, J = 7.1 Hz, 3H), 1.21 (d, J = 6.5 Hz, 6H), 1.28 (m, 2H), 1.41 (m, 2H), 1.94 (m, 2H), 2.18 (s, 3H), 2.31 (t, J = 7.4 Hz, 2H), 2.42 (t, J = 6.6 Hz, 2H), 3.06 (t, J = 7.6 Hz, 2H), 3.61 (quint, J = 6.4 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 57.34, 55.86, 52.18, 46.07, 41.79, 29.25, 24.36, 21.74, 20.62, 14.06.
- 13.
N-isopropyl-3-morpholinopropane-1-sulfonamide
3-chloropropanesulfonyl chloride (1.44 g, 8.15 mmol) was added to a round-bottom flask at 0 °C and dissolved in THF (3 mL) under argon, whereby isopropyl amine (4 mL, 0.05 mol) was added dropwise. The mixture was stirred for 20 min, after which time THF and the excess of amine were evaporated under reduced pressure. The remaining residue was dissolved in dichloromethane (10 mL) and washed with 1 M hydrochloric acid (10 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated. 1H-NMR was run on this intermediate to confirm complete conversion and used without further purification. The resulting liquid product was transferred to a pressure vessel and dissolved in toluene (4 mL). Potassium iodide (10 mg, 0.06 mmol) and morpholine (1 mL, 11.49 mmol) were added and the mixture was heated to 130 °C for 48 hrs. The resulting yellow liquid was filtered and the excess of toluene was evaporated. The resulting mixture was purified using flash column chromatography (gradient elution-ethyl acetate: hexane with an increase in ethyl acetate from 66% to 100%) to obtain a yellow liquid (0.16 g, 32%). 1H-NMR (300 MHz, CDCl3) δ 1.20 (d, J = 6.7 Hz, 6H), 1.95 (quint, J = 7.5 Hz, 2H), 2.41 (m, 6H), 3.06 (t, J = 7.7 Hz, 2H), 3.59 (m, J = 6.7 Hz, 1H), 3.67 (t, J = 4.6 Hz, 4H), 4.67 (d, J = 7.6 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 66.84, 56.76, 53.41, 51.90, 46.12, 24.34, 20.87.
- 14.
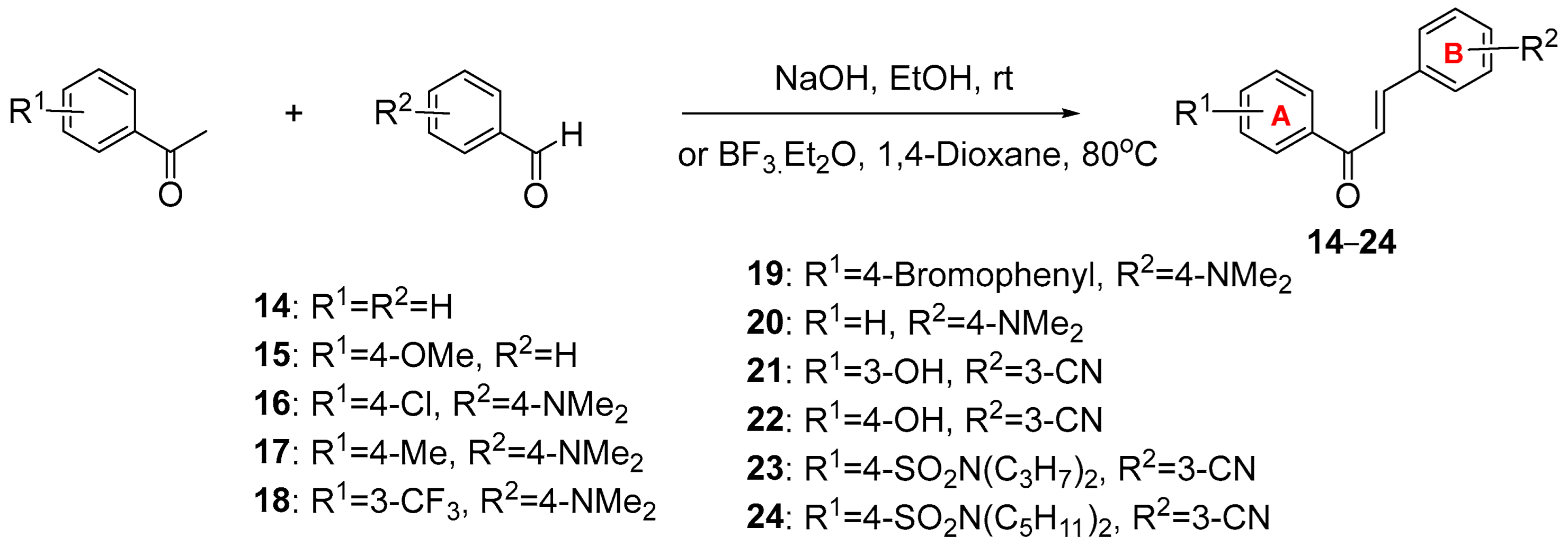
(E)-Chalcone
A solution of acetophenone (1.05 g, 8.17 mmol) in absolute ethanol (10 mL) was added to an aqueous solution of 10% NaOH (30 mL) at 0 °C. The mixture was stirred for 15 min, after which time benzaldehyde (1.0347 g, 9.75 mmol) was added. The reaction mixture was then stirred at room temperature for 24 h. The precipitated product was vacuum-filtered and washed with small portions of water/ethanol to yield the desired chalcone as a white solid (1.52 g, 75%). 1H-NMR (300 MHz, CDCl3) δ 7.42 (m, 3H), 7.54 (m, 4H), 7.65 (m, 2H), 7.82 (d, J = 15.7 Hz, 1H), 8.02 (d, J = 9 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 190.59, 144.88, 138.20, 134.87, 132.82, 130.58, 128.98, 128.65, 128.52, 128.47, 122.06. Melting point: 52.6–55.3 °C.
- 15.
(E)-1-(4-methoxyphenyl)-3-phenylprop-2-en-1-one
A solution of 1-(4-methoxyphenyl)ethan-1-one (1.02 g, 6.82 mmol) in absolute ethanol (10 mL) was added to an aqueous solution of 10% NaOH (30 mL) at 0 °C. The mixture was stirred for 15 min, after which time benzaldehyde (1.0324 g, 9.72 mmol) was added. The reaction mixture was then stirred at room temperature for 24 h. The precipitated product was vacuum-filtered and washed with small portions of water/ethanol to yield the desired chalcone as a white solid (1.87 g, 81%). 1H-NMR (300 MHz, CDCl3) δ 3.89 (s, 3H), 6.98 (d, J = 8.9 Hz, 2H), 7.42 (m, 3H), 7.55 (d, J = 15.7 Hz, 1H), 7.64 (m, 2H), 7.80 (d, J = 15.7 Hz, 1H), 8.05 (d, J = 8.9 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 188.74, 163.43, 143.99, 135.07, 130.83, 130.35, 128.93, 128.37, 125.76, 121.86, 113.85, 55.52. Melting point: 104.0–107.4 °C.
- 16.
(E)-1-(4-chlorophenyl)-3-(4-(dimethylamino) phenyl) prop-2-en-1-one
A solution of 1-(4-chlorophenyl)ethan-1-one (1.24 g, 8.03 mmol) in absolute ethanol (10 mL) was added to an aqueous solution of 10% NaOH (30 mL) at 0 °C. The mixture was stirred for 15 min, after which time 4-(dimethylamino)benzaldehyde (1.0524 g, 7.05 mmol) was added. The reaction mixture was then stirred at room temperature for 24 h. The precipitated product was vacuum-filtered and washed with small portions of water/ethanol to yield the desired chalcone as an orange solid (1.59 g, 79%). 1H-NMR (300 MHz, CDCl3) δ 3.05 (s, 6H), 6.69 (d, J = 8.9 Hz, 2H), 7.29 (d, J = 15.4 Hz, 1H), 7.45 (d, J = 8.6 Hz, 2H), 7.5 (d, J = 8.9 Hz, 2H), 7.80 (d, J = 15.4 Hz, 1H), 7.95 (d, J = 8.6 Hz, 1H). 13C-NMR (75 MHz, CDCl3) δ 188.74, 163.43, 143.99, 135.07, 130.83, 130.35, 128.93, 128.37, 125.76, 121.86, 113.85, 55.52. Anal. Calcd for C17H16ClNO: C, 71.45; H, 5.64; Cl, 12.41; N, 4.90. Found: C, 71.44; H, 5.68; Cl, 12.21; N, 4.98. Melting point: 137.9–141.9 °C.
- 17.
(E)-3-(4-(dimethylamino)phenyl)-1-(p-tolyl)prop-2-en-1-one
A solution of 1-(p-tolyl)ethan-1-one (1.37 g, 10.2 mmol) in absolute ethanol (10 mL) was added to an aqueous solution of 10% NaOH (30 mL) at 0°C. The mixture was stirred for 15 min, after which time 4-(dimethylamino)benzaldehyde (1.1548 g, 7.74 mmol) was added. The reaction mixture was then stirred at room temperature for 24 h. The precipitated product was vacuum-filtered and washed with small portions of water/ethanol to yield the desired chalcone as an orange solid (1.44 g, 70%). 1H-NMR (300 MHz, CDCl3) δ 2.42 (s, 3H), 3.04 (s, 6H), 6.77 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 7.8 Hz, 2H), 7.35 (d, J = 15.5 Hz, 1H), 7.55 (d, J = 8.3 Hz, 2H), 7.78 (d, J = 15.5 Hz, 1H), 7.92 (d, J = 7.6 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 190.20, 145.38, 142.89, 136.40, 130.35, 129.17, 128.46, 116.94, 111.87, 40.21, 21.66. Anal. Calcd for C18H19NO: C, 81.47; H, 7.22; N, 5.28. Found: C, 81.70; H, 7.17; N, 5.26. Melting point: 118.6–120.9 °C.
- 18.
(E)-3-(4-(dimethylamino)phenyl)-1-(3-(trifluoromethyl)phenyl)prop-2-en-1-one
A solution of 1-(3-(trifluoromethyl) phenyl)ethan-1-one (1.08 g, 5.74 mmol) in absolute ethanol (10 mL) was added to an aqueous solution of 10% NaOH (30 mL) at 0 °C. The mixture was stirred for 15 min, after which time 4-(dimethylamino)benzaldehyde (1.0043 g, 6.73 mmol) was added. The reaction mixture was then stirred at room temperature for 24 h. The precipitated product was vacuum-filtered and washed with small portions of water/ethanol to yield the desired chalcone as an orange solid (1.42 g, 66%). 1H-NMR (300 MHz, CDCl3) δ 3.06 (s, 6H), 6.70 (d, J = 8.9 Hz, 2H), 7.30 (d, J = 15.4 Hz, 1H), 7.61 (m, 4H), 7.83 (m, 2H), 8.20 (m, 2H). 13C-NMR (75 MHz, CDCl3) δ 189.05, 152.29, 147.10, 139.67, 131.46, 130.75, 129.08, 128.55, 128.50, 125.12, 125.07, 122.19, 115.75, 111.78, 40.12. Anal. Calcd for C18H16F3NO: C, 67.70; H, 5.05; F, 17.85; N, 4.39. Found: C, 66.94; H, 4.96; F, 17.87; N, 4.22. Melting point: 85.2–86.4 °C.
- 19.
(E)-1-(4’-bromo-[1,1’-biphenyl]-4-yl)-3-(4-(dimethylamino)phenyl)prop-2-en-1-one
A solution of 1-(4’-bromo-[1,1’-biphenyl]-4-yl)ethan-1-one (1.03 g, 3.74 mmol) in absolute ethanol (10 mL) was added to an aqueous solution of 10% NaOH (30 mL) at 0 °C. The mixture was stirred for 15 min, after which time 4-(dimethylamino)benzaldehyde (1.1178 g, 7.49 mmol) was added. The reaction mixture was then stirred at room temperature for 24 h. The precipitated product was vacuum-filtered and washed with small portions of water/ethanol to yield the desired chalcone as a yellow solid (2.25 g, 74%). 1H-NMR (300 MHz, CDCl3) δ 3.06 (s, 6H), 6.70 (d, J = 7.5 Hz, 2H), 7.36 (d, J = 15.4 Hz, 1H), 7.59 (m, 8H), 7.84 (d, J = 15.3 Hz, 1H), 8.09 (d, J = 6.9 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 189.91, 152.08, 145.97, 143.54, 139.06, 138.12, 132.05, 130.50, 129.02, 128.83, 126.93, 122.59, 122.42, 116.65, 111.82, 40.15. Anal. Calcd for C23H20BrNO: C, 67.99; H, 4.96; Br, 19.67; N, 3.45. Found: C, 67.68; H, 5.05; Br, 19.46; N, 3.39. Melting point: 183.2–187.6 °C.
- 20.
(E)-3-(4-(dimethylamino)phenyl)-1-phenylprop-2-en-1-one
A solution of acetophenone (1.08 g, 9.01 mmol) in absolute ethanol (10 mL) was added to an aqueous solution of 10% NaOH (30 mL) at 0 °C. The mixture was stirred for 15 min, after which time 4-(dimethylamino)benzaldehyde (0.9784 g, 6.55 mmol) was added. The reaction mixture was then stirred at room temperature for 24 h. The precipitated product was vacuum-filtered and washed with small portions of water/ethanol to yield the desired chalcone as an orange solid (1.33 g, 81%). 1H-NMR (300 MHz, CDCl3) δ 3.05 (s, 6H), 6.70 (d, J = 8.8 Hz, 2H), 7.33 (d, J = 15.5 Hz, 1H) 7.52 (m, 5H), 7.79 (d, J = 15.5 Hz, 1H), 8.00 (d, J = 6.9 Hz, 2H). 13C-NMR (75 MHz, CDCl3) δ 190.72, 152.02, 145.89, 139.08, 139.06, 130.44, 128.47, 128.32, 122.61, 116.87, 111.82, 40.16. Anal. Calcd for C17H17NO: C, 81.24; H, 6.82; N, 5.57. Found: C, 80.81; H, 6.82; N, 5.50. Melting point: 106.9–110.3 °C.
- 21.
(E)-3-(3-(3-hydroxyphenyl)-3-oxoprop-1-en-1-yl)benzonitrile
Boron trifluoride etherate (48% BF3, 781 mg, 5.5 mmol) was added to a stirred solution of 3′-hydroxyacetophenone (150 mg, 1.1 mmol) and 3-cyanobenzaldehyde (289 mg, 2.2 mmol) in 1,4-dioxane (10 mL), and the reaction mixture was heated at 80 °C for 14–24 h. After cooling, the resultant solution was partitioned with EtOAc, washed with 10% HCl (aq), distilled water, and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified using column chromatography (n-hexane:EtOAc = 3:1, 1:1) to obtain 21 as a solid (114 mg, 41%). 1H-NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 8.50 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 8.04 (d, J = 15.8 Hz, 1H), 7.90 (dt, J = 7.8, 1.3 Hz, 1H), 7.73 (dt, J = 15.7 Hz, 1H), 7.70–7.63 (m, 2H), 7.51–7.48 (m, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.08 (ddd, J = 8.1, 2.6, 1.0 Hz, 1H).
- 22.
(E)-3-(3-(4-hydroxyphenyl)-3-oxoprop-1-en-1-yl)benzonitrile
The procedure applied to the synthesis of
21 was used with boron trifluoride etherate (48% BF
3, 781 mg, 5.5 mmol), 4-hydroxy acetophenone (150 mg, 1.1 mmol) and 3-cyanobenzaldehyde (289 mg, 2.2 mmol) to obtain
22 as a yellow solid (92 mg, 34%) (
Scheme 4).
1H-NMR (400 MHz, DMSO-
d6) δ 10.48 (s, 1H), 8.48 (s, 1H), 8.17 (d,
J = 8.0 Hz, 1H), 8.13–8.06 (m,
3H), 7.88 (dt,
J = 8.0, 1.6 Hz, 1H), 7.69 (d,
J = 15.6 Hz, 1H), 7.66 (t,
J = 8.0 Hz, 1H), 6.93–6.89 (m, 2H).
- 23.
(23a) 4-Acetyl-N,N-dipropylbenzenesulfonamide
A mixture of 4-acetylbenzenesulfonyl chloride (300 mg, 1.37 mmol), dipropylamine (151 mg, 1.50 mmol), and triethylamine (277 mg, 2.74 mmol) in anhydrous THF (10 mL) was stirred at room temperature overnight. Water was added, and the reaction mixture was extracted with EtOAc (3 times). The combined organic layer was washed with brine (100 mL), dried over Na2SO4, and filtered. The removal of solvent in vacuo presented as yellow oil (154 mg, 40%).
(23) (E)-4-(3-(3-cyanophenyl)acryloyl)-N,N-dipropylbenzenesulfonamide
The procedure applied to the synthesis of 21 was used with boron trifluoride etherate (48% BF3, 325 mg, 2.29 mmol), 23a (130 mg, 0.46 mmol), and 3-cyanobenzaldehyde (120 mg, 0.92 mmol) to obtain 23 as an ivory fluffy solid (81 mg, 45%) after purification by column chromatography (n-hexane:EtOAc = 10:1, 5:1). 1H-NMR (400 MHz, DMSO-d6) δ 8.52 (s, 1H), 8.35 (d, J = 8.6 Hz, 2H), 8.22 (d, J = 8.0 Hz, 1H), 8.13 (d, J = 15.7 Hz, 1H), 7.99 (d, J = 8.5 Hz, 2H), 7.92 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 15.7 Hz, 1H), 7.69 (t, J = 7.7 Hz, 1H), 3.09 (d, J = 7.6 Hz, 4H), 1.49 (sextet, J = 7.6 Hz, 4H), 0.82 (t, J = 7.6 Hz, 6H).
- 24.
(24a) 4-Acetyl-N,N-dipentylbenzenesulfonamide
The procedure applied to the synthesis of 23a was used with 4-acetylbenzenesulfonyl chloride (300 mg, 1.37 mmol), diamylamine (235 mg, 1.50 mmol), and triethylamine (277 mg, 2.74 mmol) to obtain 24a as yellow oil (187 mg, 40%).
(24) (E)-4-(3-(3-cyanophenyl)acryloyl)-N,N-dipentylbenzenesulfonamide
The procedure applied to the synthesis of 21 was used with boron trifluoride etherate (48% BF3, 271 mg, 1.91 mmol), 24a (130 mg, 0.38 mmol), and 3-cyanobenzaldehyde (100 mg, 0.77 mmol) to obtain 24 as an off-white crystal (37 mg, 21%) after purification by column chromatography (n-hexane:EtOAc = 10:1). 1H-NMR (400 MHz, DMSO-d6) δ 8.51 (s, 1H), 8.35 (d, J = 8.6 Hz, 2H), 8.22 (d, J = 7.6 Hz, 1H), 8.13 (d, J = 15.7 Hz, 1H), 7.97 (d, J = 8.8 Hz, 2H), 7.92 (dt, J = 7.7, 1.2 Hz, 1H), 7.81 (d, J = 15.7 Hz, 1H), 7.69 (t, J = 7.7 Hz, 1H), 3.11 (t, J = 7.6 Hz, 4H), 1.46 (quintet, J = 7.6 Hz, 4H), 1.29–1.18 (m, 8 H), 0.84 (t, J = 7.2 Hz, 6H).
- 25.
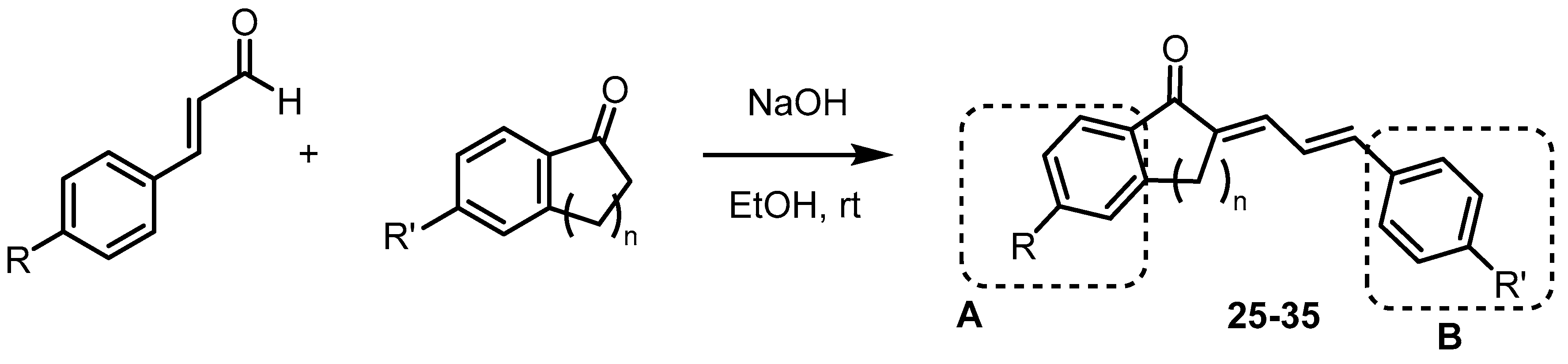
(2E,4E)-1-(4-methoxyphenyl)-5-phenylpenta-2,4-dien-1-one
4′-methoxyacetophenone (200 mg, 1.33 mmol) and trans-cinnamaldehyde (0.168 mL, 1.33 mmol) were dissolved in absolute ethanol (10 mL) at room temperature. To this stirring solution, 6M NaOH (1 mL) was added dropwise. Precipitate formed instantaneously and the mixture was stirred for 15 min at room temperature. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as a bright yellow powder (321 mg, 91%). 1H-NMR (300 MHz, Chloroform-d) δ 8.00 (d, J = 8.0 Hz, 2H), 7.69–7.53 (m, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.42–7.27 (m, 3H), 7.11 (d, J = 14.5 Hz, 1H), 7.04–6.88 (m, 4H), 3.86 (s, 3H). 13C-NMR (75 MHz, Chloroform-d) δ 188.66, 163.33, 144.04, 141.43, 136.17, 131.06, 130.70, 129.12, 128.84, 127.25, 127.04, 125.20, 113.81, 55.48. Melting point: 73–76 °C.
- 26.
(2E,4E)-1-(4-chlorophenyl)-5-(4-(dimethylamino)phenyl)penta-2,4-dien-1-one
4′-chloroacetophenone (0.250 mL, 1.92 mmol), and 4-(dimethylamino)cinnamaldehyde (305 mg, 1.75 mmol) were dissolved in absolute ethanol (15 mL) and THF (1 mL) at 50 °C. The solution was slowly cooled to room temperature, and 6M NaOH (1 mL) was added dropwise during this time. Precipitate slowly formed, and the reaction mixture was stirred for 1 h at room temperature. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as an orange powder (486 mg, 81%). 1H-NMR (300 MHz, Chloroform-d) δ 7.91 (d, J = 8.5 Hz, 2H), 7.63 (dd, J = 14.7, 10.9 Hz, 1H), 7.44 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 8.9 Hz, 3H), 7.04–6.77 (m, 3H), 6.68 (d, J = 8.9 Hz, 2H), 3.02 (s, 6H). 13C-NMR (75 MHz, Chloroform-d) δ 189.07, 151.14, 146.96, 143.74, 138.55, 137.00, 129.70, 129.05, 128.76, 123.96, 122.17, 121.82, 111.96, 40.22. Melting point: 161–164 °C.
- 27.
(2E,4E)-5-(4-(dimethylamino)phenyl)-1-(p-tolyl)penta-2,4-dien-1-one
4′-methylacetophenone (0.250 mL, 1.88 mmol) and 4-(dimethylamino)cinnamaldehyde (300 mg, 1.71 mmol) were dissolved in absolute ethanol (15 mL) and THF (1 mL) at 50 °C. The solution was slowly cooled to room temperature, and 6M NaOH (1 mL) was added dropwise during this time. Precipitate slowly formed, and the reaction mixture was stirred for 1 h at room temperature. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as a bright red powder (351 mg, 71%). 1H-NMR (300 MHz, Chloroform-d) δ 7.89 (d, J = 8.2 Hz, 2H), 7.63 (dd, J = 14.8, 10.6 Hz, 1H), 7.40 (d, J = 8.9 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 7.00–6.87 (m, 3H), 6.68 (d, J = 8.9 Hz, 2H), 3.02 (s, 6H), 2.42 (s, 3H). 13C-NMR (75 MHz, Chloroform-d) δ 190.06, 151.03, 145.96, 143.01, 142.81, 136.08, 129.18, 128.85, 128.42, 124.22, 122.64, 122.51, 112.00, 40.23, 21.66. Melting point: 158–160 °C.
- 28.
(2E,4E)-1-(4-chlorophenyl)-5-phenylpenta-2,4-dien-1-one
To a stirring solution of 4′-chloroacetophenone (0.250 mL, 1.92 mmol) and trans-cinnamaldehyde (0.250 mL, 1.99 mmol), in absolute ethanol (10 mL), 6 M NaOH (1 mL) was added dropwise at room temperature. Precipitate formed instantaneously and the reaction mixture was stirred for an additional 15 min at room temperature. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as a yellow green powder (445 mg, 95%). 1H-NMR (300 MHz, Chloroform-d) δ 7.92 (d, J = 8.6 Hz, 2H), 7.69–7.55 (m, 1H), 7.54–7.43 (m, 4H), 7.37 (m, 3H), 7.10–6.91 (m, 3H). 13C-NMR (75 MHz, Chloroform-d) δ 189.09, 145.34, 142.43, 139.06, 136.48, 135.96, 129.79, 129.78, 129.38, 128.89, 127.35, 126.74, 124.74. Melting point: 138–139 °C.
- 29.
(E)-2-((E)-3-(4-(dimethylamino)phenyl)allylidene)-2,3-dihydro-1H-inden-1-one
1-indanone (200 mg, 1.51 mmol) and 4-(dimethylamino)cinnamaldehyde (240 mg, 1.38 mmol) were dissolved in absolute ethanol (15 mL) and THF (1 mL) at 50 °C. The solution was slowly cooled to room temperature, and 6M NaOH (1 mL) was added dropwise during this time. Precipitate slowly formed, and the reaction mixture was stirred for 1 h at room temperature. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as an orange powder (327 mg, 82%). 1H-NMR (300 MHz, Chloroform-d) δ 7.87 (d, J = 7.6 Hz, 1H), 7.56 (m, 2H), 7.48–7.35 (m, 4H), 6.99 (d, J = 15.3 Hz, 1H), 6.84 (dd, J = 15.2, 11.4 Hz,10H), 6.68 (d, J = 8.8 Hz, 2H), 3.82 (s, 2H), 3.02 (s, 6H). 13C-NMR (75 MHz, Chloroform-d) δ 193.63, 151.06, 148.85, 143.14, 139.74, 134.96, 133.89, 133.35, 128.93, 127.34, 126.12, 124.43, 123.96, 119.81, 111.99, 40.22, 30.54. Melting point: 168–170 °C.
- 30.
(E)-6-methoxy-2-((E)-3-(4-methoxyphenyl)allylidene)-3,4-dihydronaphthalen-1(2H)-one
6-methoxy-1-tetralone (250 mg, 1.42 mmol) and 4-methoxycinnamaldehyde (230 mg, 1.42 mmol) were dissolved in absolute ethanol (10 mL) at room temperature and 6M NaOH (1 mL) was added dropwise. Precipitate slowly formed, and the reaction mixture was stirred for 30 min at room temperature. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as a bright yellow powder (248 mg, 55%). 1H-NMR (300 MHz, Chloroform-d) δ 8.09 (d, J = 8.7 Hz, 1H), 7.52 (d, J = 8.6 Hz, 1H), 7.46 (d, J = 8.6 Hz, 2H), 7.10–6.79 (m, 5H), 6.72 (s, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 2.98 (s, 4H). 13C-NMR (75 MHz, Chloroform-d) δ 186.26, 163.28, 160.21, 145.84, 140.28, 135.83, 133.36, 130.52, 129.55, 128.57, 127.45, 121.49, 114.23, 113.15, 112.33, 55.44, 55.36, 29.19, 25.99. Melting point: 139–140 °C.
- 31.
(2E,4E)-5-(4-chlorophenyl)-1-(4-nitrophenyl)penta-2,4-dien-1-one
4′-nitroacetophenone (161 mg, 0.972 mmol) and 4-chlorocinnamaldehyde (162 mg, 0.972 mmol) were dissolved in absolute ethanol (15 mL) at 50 °C. The solution was slowly cooled to room temperature, and 6 M NaOH (1 mL) was added dropwise during this time. Precipitate formed instantaneously, and the reaction mixture was stirred at room temperature for 15 min. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as a yellow green powder (252 mg, 83%). 1H-NMR (300 MHz, Chloroform-d) δ 8.34 (d, J = 8.9 Hz, 2H), 8.10 (d, J = 9.0 Hz, 2H), 7.62 (ddd, J = 14.9, 6.5, 3.9 Hz, 1H), 7.45 (d, J = 8.7 Hz, 2H), 7.36 (d, J = 8.7 Hz, 2H), 7.11–6.97 (m, 3H). 13C-NMR (75 MHz, Chloroform-d) δ 188.77, 149.97, 146.29, 142.93, 141.99, 135.44, 134.22, 129.29, 129.20, 128.60, 126.95, 124.81, 123.87. Melting point: 122–123 °C.
- 32.
(E)-2-((E)-3-(4-(dimethylamino)phenyl)allylidene)-2,3-dihydro-1H-inden-1-one
1,3-indandione (200 mg, 1.37 mmol) and trans-cinnamaldehyde (0.190 mL, 1.51 mmol) were dissolved in absolute ethanol (15 mL) at room temperature, and 6 M NaOH (1 mL) was added dropwise. The reaction mixture was stirred at room temperature overnight, then cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as a yellow powder (90 mg, 25%). 1H-NMR (300 MHz, Chloroform-d) δ 8.45 (dd, J = 15.5, 12.0 Hz, 1H), 8.03–7.91 (m, 2H), 7.84–7.75 (m, 2H), 7.72–7.59 (m, 3H), 7.43 (m, 3H), 7.34 (d, J = 15.5 Hz, 1H). 13C-NMR (75 MHz, Chloroform-d) δ 151.16, 144.68, 142.14, 135.51, 135.15, 135.03, 130.94, 129.05, 128.70, 123.62, 123.14, 122.97. Melting point: 151–152 °C.
- 33.
(E)-2-((E)-3-(2-nitrophenyl)allylidene)-3,4-dihydronaphthalen-1(2H)-one
1-tetralone (0.210 mL, 1.58 mmol) and 2-nitrocinnamaldehyde (250 mg, 1.41 mmol) were dissolved in absolute ethanol (20 mL) and THF (2 mL) at 50 °C. The solution was slowly cooled to room temperature, and 6 M NaOH (1 mL) was added dropwise during this time. Precipitate formed instantaneously and the reaction mixture was stirred for 15 min at room temperature. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as a yellow powder (260 mg, 60%). 1H-NMR (300 MHz, Chloroform-d) δ 7.86 (d, J = 7.8 Hz, 1H), 7.76 (d, J = 8.1 Hz, 1H), 7.59 (d, J = 7.9 Hz, 1H), 7.44 (t, J = 7.9 Hz, 1H), 7.35–7.21 (m, 4H), 7.15 (t, J = 7.5 Hz, 1H), 7.08 (d, J = 7.6 Hz, 1H), 6.95 (dd, J = 15.1, 11.7 Hz, 1H), 2.83 (s, 6H). 13C-NMR (75 MHz, Chloroform-d) δ 186.93, 147.71, 143.29, 136.61, 134.59, 134.40, 133.25, 133.21, 133.16, 131.87, 128.95, 128.30, 128.22, 127.96, 127.83, 126.90, 124.69, 28.46, 26.02. Melting point: 188–189 °C.
- 34.
(2E,4E)-5-(4-(dimethylamino)phenyl)-1-phenylpenta-2,4-dien-1-one
Acetophenone (0.200 mL, 1.71 mmol) and 4-(dimethylamino)cinnamaldehyde (275 mg, 1.56 mmol) were dissolved in absolute ethanol (10 mL) and THF (1 mL) at 50 °C. The solution was slowly cooled to room temperature, and 6 M NaOH (1 mL) was added dropwise during this time. Precipitate slowly formed, and the reaction mixture was stirred for 1 h at room temperature. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as a red flaky solid (327 mg, 76%). 1H-NMR (300 MHz, Chloroform-d) δ 7.98 (d, J = 6.8 Hz, 2H), 7.64 (dd, J = 14.8, 10.6 Hz, 1H), 7.58–7.44 (m, 3H), 7.40 (d, J = 8.9 Hz, 2H), 7.04–6.78 (m, 3H), 6.68 (d, J = 8.9 Hz, 2H), 3.01 (s, 6H). 13C-NMR (75 MHz, Chloroform-d) δ 190.55, 151.09, 146.45, 143.18, 138.71, 132.28, 128.94, 128.48, 128.29, 124.13, 122.58, 122.41, 112.00, 40.22. Melting point: 153–154 °C.
- 35.
(E)-2-((E)-3-(4-(dimethylamino) phenyl)allylidene)-3,4-dihydronaphthalen-1(2H)-one
1-tetralone (0.200 mL, 1.50 mmol) and 4-(dimethylamino)cinnamaldehyde (240 mg, 1.36 mmol) were dissolved in absolute ethanol (15 mL) and THF (1 mL) at 50 °C. The solution was slowly cooled to room temperature, and 6 M NaOH (1 mL) was added dropwise during this time. Precipitate slowly formed, and the reaction mixture was stirred for 1 h at room temperature. A few chips of ice were added, and the reaction mixture was cooled in an ice bath for 15 min. The precipitate was vacuum-filtered and washed with small portions of cold water/ethanol solution to yield the desired chalcone as a red powder (191 mg, 46%). 1H-NMR (300 MHz, Chloroform-d) δ 8.11 (d, J = 7.7 Hz, 1H), 7.60 (dd, J = 8.1, 2.3 Hz, 1H), 7.44 (m, 3H), 7.35 (t, J = 7.5 Hz, 1H), 7.25 (d, J = 6.5 Hz, 1H), 7.05–6.85 (m, 2H), 6.68 (d, J = 8.7 Hz, 3H), 3.02 (s, 6H), 3.00 (s, 4H). 13C-NMR (75 MHz, Chloroform-d) δ 187.25, 150.88, 143.36, 142.10, 137.61, 134.16, 132.68, 131.51, 128.72, 128.06, 127.97, 126.86, 124.77, 119.12, 112.04, 40.26, 28.79, 25.77. Melting point: 145–147 °C softening, 275–285 °C melting.



 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
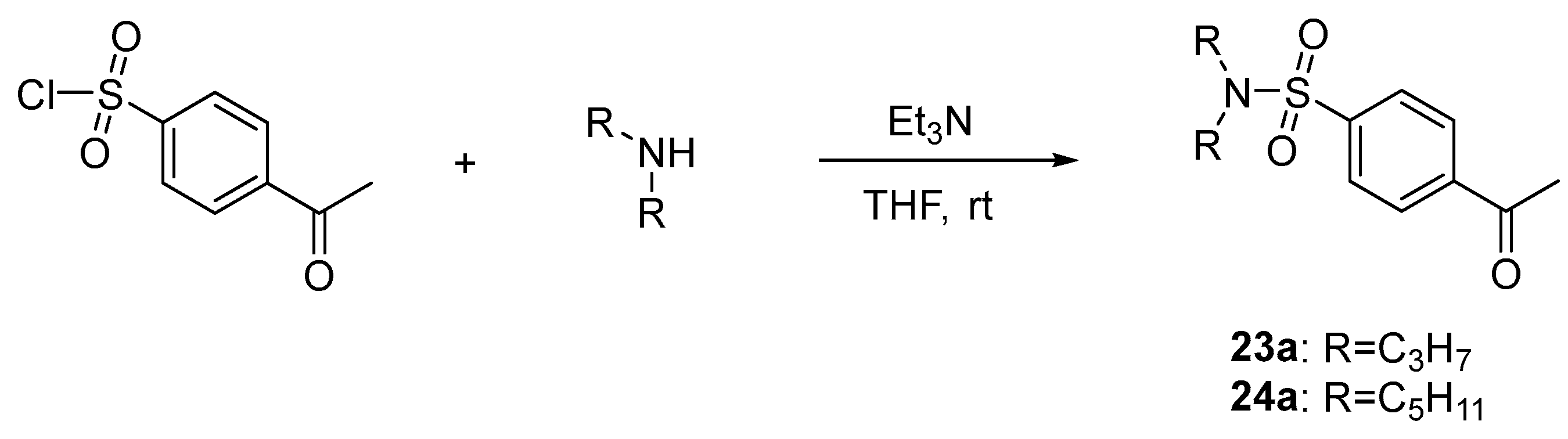{kind=link}


























































