
Tetrasomy 9p, a Prenatal Challenge: Two Novel Cases



Abstract
:1. Introduction
2. Case Presentation
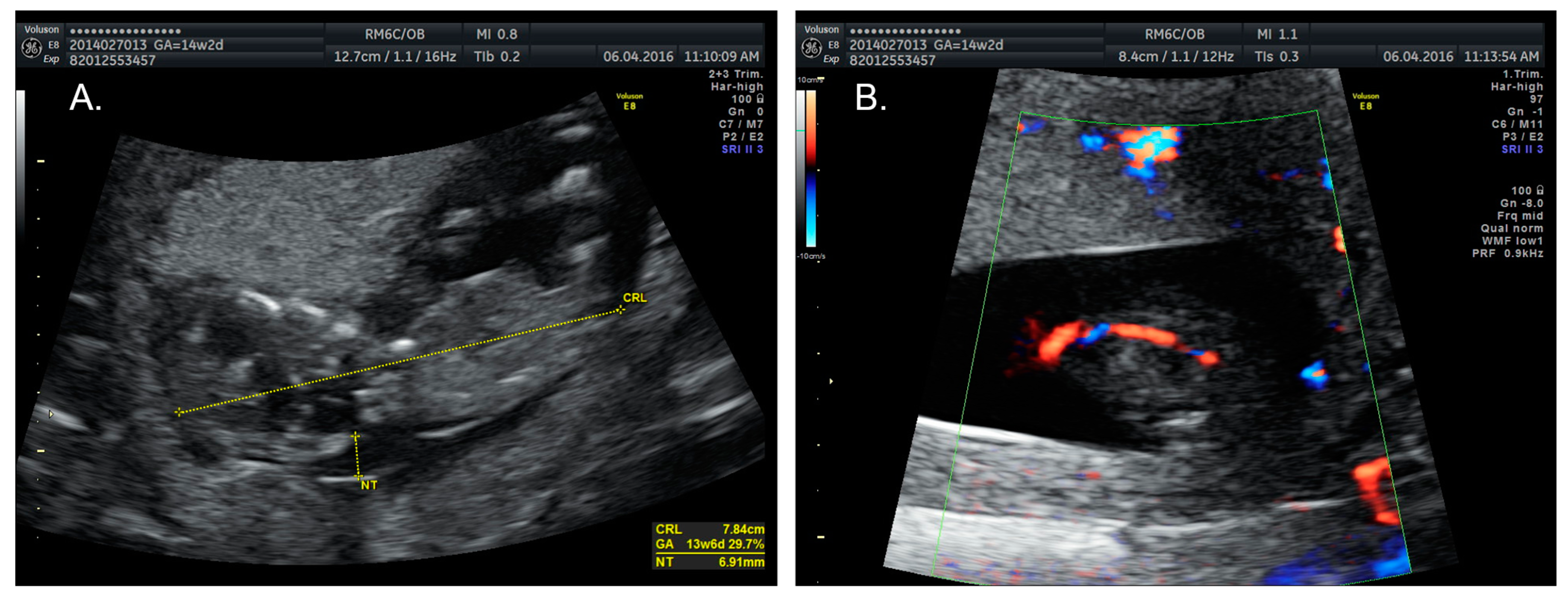
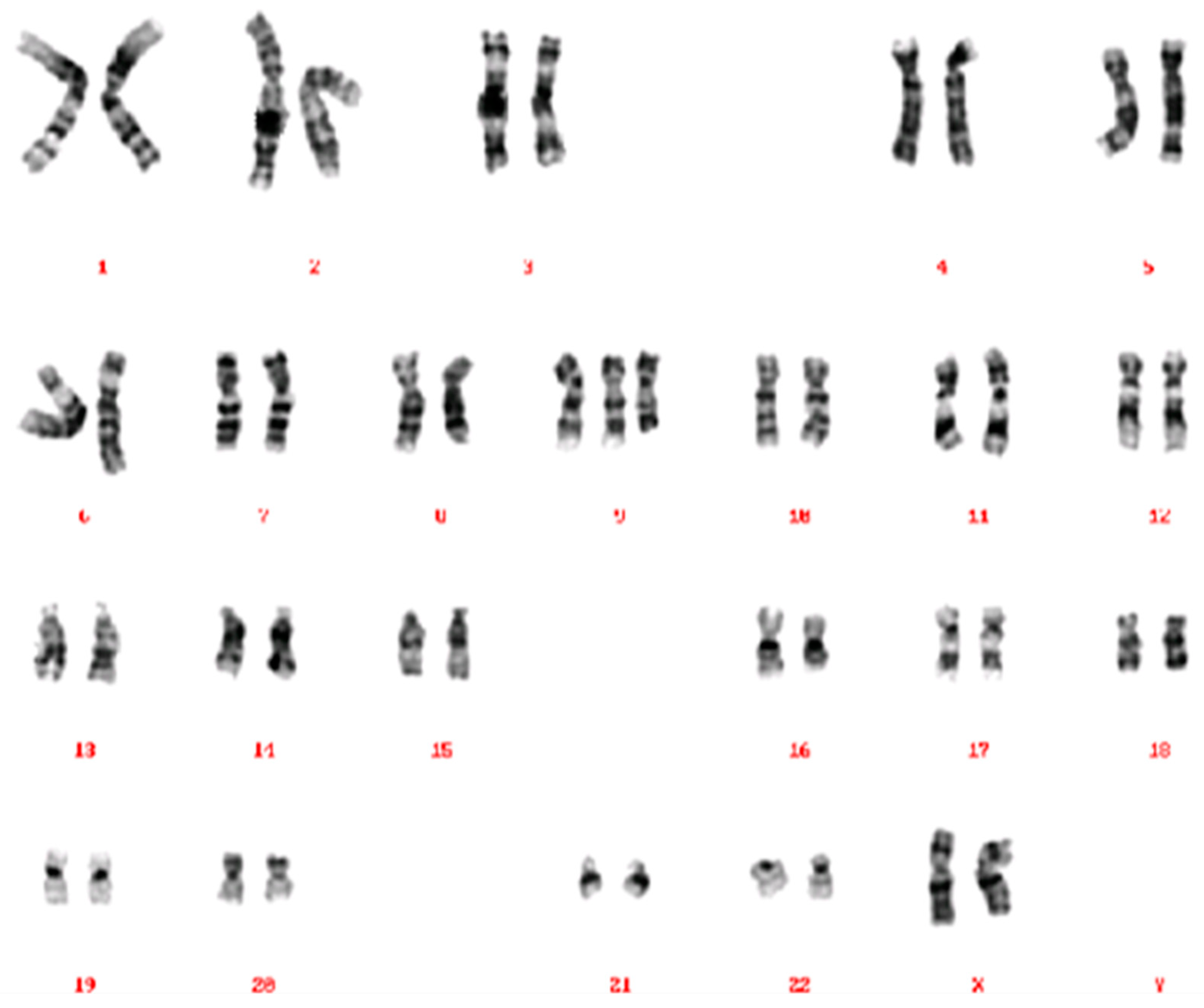
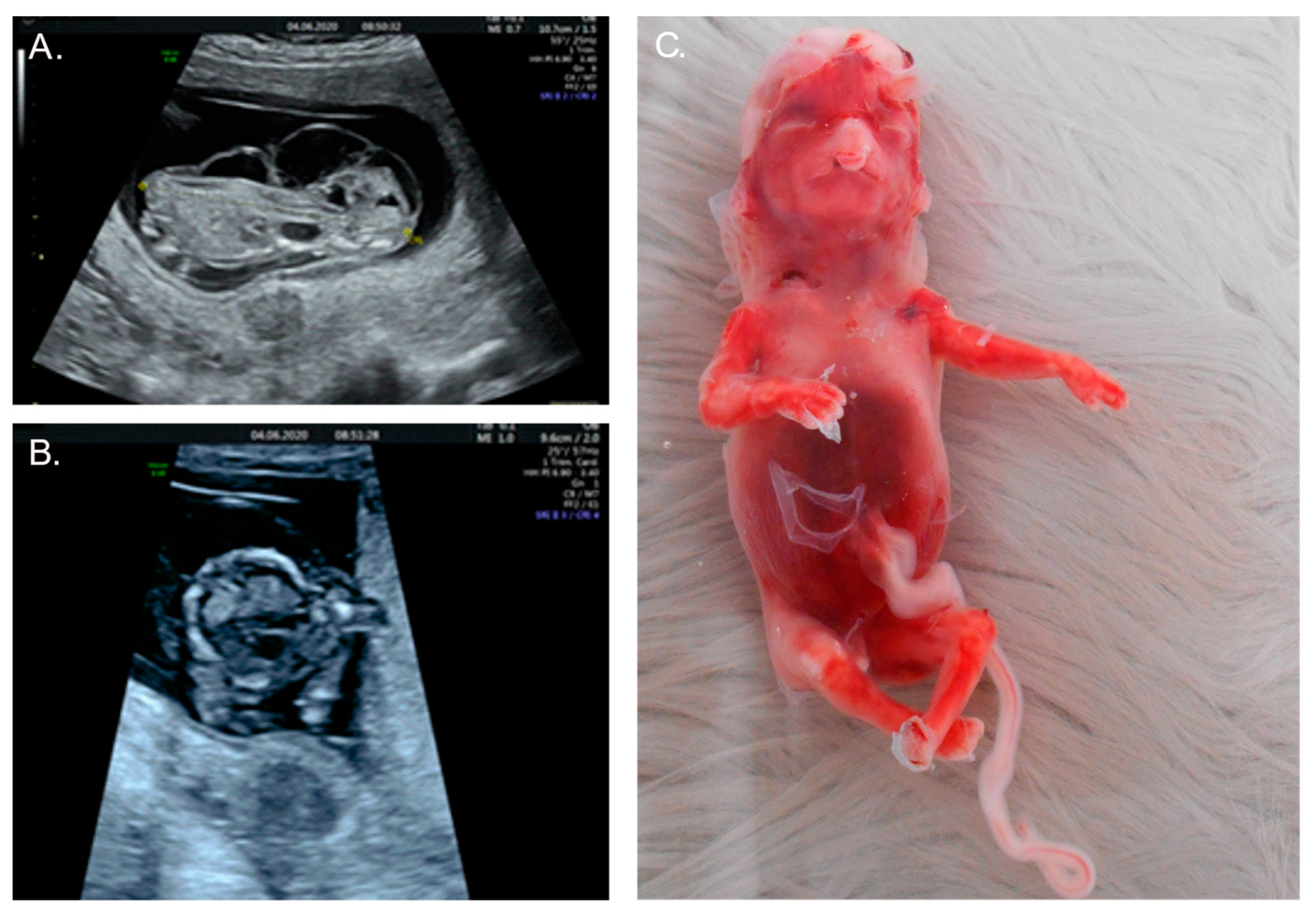
2.1. Case 1
2.2. Case 2
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El Khattabi, L.; Jaillard, S.; Andrieux, J.; Pasquier, L.; Perrin, L.; Capri, Y.; Benmansour, A.; Toutain, A.; Marcorelles, P.; Vincent-Delorme, C.; et al. Clinical and molecular delineation of Tetrasomy 9p syndrome: Report of 12 new cases and literature review. Am. J. Med. Genet. Part A 2015, 167, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Ghymers, D.; Hermann, B.; Distèche, C.; Frederic, J. Partial tetrasomy of number 9 chromosome, and mosaicism in a child with multiple malformations (author’s transl). Humangenetik 1973, 20, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Vinkšel, M.; Volk, M.; Peterlin, B.; Lovrecic, L. A systematic clinical review of prenatally diagnosed tetrasomy 9p. Balk. J. Med. Genet. 2019, 22, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liehr, T.; Al-Rikabi, A. Mosaicism: Reason for normal phenotypes in carriers of small supernumerary marker chromosomes with known adverse outcome. A systematic review. Front. Genet. 2019, 10, 1131. [Google Scholar] [CrossRef]
- Nakamura-Pereira, M.; Cima, L.C.; Llerena, J.C., Jr.; Guerra, F.A.; Peixoto-Filho, F.M. Sonographic findings in a case of tetrasomy 9p associated with increased nuchal translucency and Dandy-Walker malformation. J. Clin. Ultrasound 2009, 37, 471–474. [Google Scholar] [CrossRef]
- Henriques-Coelho, T.; Oliva-Teles, N.; Fonseca-Silva, M.L.; Tibboel, D.; Guimarães, H.; Correia-Pinto, J. Congenital diaphragmatic hernia in a patient with tetrasomy 9p. J. Pediatr. Surg. 2005, 40, e29–e31. [Google Scholar] [CrossRef] [Green Version]
- Lazebnik, N.; Cohen, L. Prenatal diagnosis and findings of tetrasomy 9p. J. Obstet. Gynaecol. Res. 2015, 41, 997–1002. [Google Scholar] [CrossRef]
- Pellestor, F. Maternal age and chromosomal abnormalities in human oocytes. Med. Sci. M/S 2004, 20, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Eichenlaub-Ritter, U. Parental age-related aneuploidy in human germ cells and offspring: A story of past and present. Environ. Mol. Mutagen. 1996, 28, 211–236. [Google Scholar] [CrossRef]
- Nicolaides, K.H. Screening for fetal aneuploidies at 11 to 13 weeks. Prenat. Diagn. 2011, 31, 7–15. [Google Scholar] [CrossRef]
- De Azevedo Moreira, L.M.; Freitas, L.M.; Gusmão, F.A.; Riegel, M. New case of non-mosaic tetrasomy 9p in a severely polymalformed newborn girl. Birth Defects Res. Part A Clin. Mol. Teratol. 2003, 67, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Dhandha, S.; Hogge, W.A.; Surti, U.; McPherson, E. Three cases of tetrasomy 9p. Am. J. Med. Genet. 2002, 113, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, R.; Saltzman, D.; Auerbach, M.; Roman, A.S. Absent nasal bone as a marker of tetrasomy 9p. Prenat. Diagn. 2011, 31, 1313. [Google Scholar] [CrossRef] [PubMed]
- McDowall, A.A.; Blunt, S.; Berry, A.C.; Fensom, A.H. Prenatal diagnosis of a case of tetrasomy 9p. Prenat. Diagn 1989, 9, 809–811. [Google Scholar] [CrossRef]
- Schaefer, G.B.; Domek, D.B.; Morgan, M.A.; Muneer, R.S.; Johnson, S.F. Tetrasomy of the short arm of chromosome 9: Prenatal diagnosis and further delineation of the phenotype. Am. J. Med. Genet. 1991, 38, 612–615. [Google Scholar] [CrossRef]
- Van Hove, J.; Kleczkowska, A.; De Bruyn, M.; Bekaert, J.; Fryns, J.P. Tetrasomy 9p: Prenatal diagnosis and fetopathological findings in a second trimester male fetus. Ann. Genet. 1994, 37, 139–142. [Google Scholar]
- Cazorla Calleja, M.R.; Verdú, A.; Félix, V. Dandy-Walker malformation in an infant with tetrasomy 9p. Brain Dev. 2003, 25, 220–223. [Google Scholar] [CrossRef]
- Deurloo, K.L.; Cobben, J.M.; Heins, Y.M.; de Ru, M.; Wijnaendts, L.C.; van Vugt, J.M. Prenatal diagnosis of tetrasomy 9p in a 19-week-old fetus with Dandy-Walker malformation: A case report. Prenat. Diagn. 2004, 24, 796–798. [Google Scholar] [CrossRef]
- Tang, W.; Boyd, B.K.; Hummel, M.; Wenger, S.L. Prenatal diagnosis of tetrasomy 9p. Am. J. Med. Genet. A 2004, 126A, 328. [Google Scholar] [CrossRef]
- Hengstschläger, M.; Bettelheim, D.; Drahonsky, R.; Repa, C.; Deutinger, J.; Bernaschek, G. Prenatal diagnosis of tetrasomy 9p with Dandy-Walker malformation. Prenat. Diagn. 2004, 24, 623–626. [Google Scholar] [CrossRef]
- Di Vera, E.; Liberati, M.; Celentano, C.; Calabrese, G.; Guanciali-Franchi, P.E.; Morizio, E.; Rotmensch, S. Rhombencephalosynapsis in a severely polymalformed fetus with non-mosaic tetrasomy 9p, in intracytoplasmic-sperm-injection pregnancy. J. Assist. Reprod. Genet. 2008, 25, 577–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutly, F.; Balmer, D.; Baumer, A.; Binkert, F.; Schinzel, A. Isochromosomes 12p and 9p: Parental origin and possible mechanisms of formation. Eur. J. Hum. Genet. 1998, 6, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.Q.; Chen, X.M.; Hu, L.; Guan, X.Y.; Lu, G.X. Prenatal diagnosis of nonmosaic tetrasomy 9p by microdissection and FISH: Case report. Chin. Med. J. 2007, 120, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Wang, L.K.; Chern, S.R.; Wu, P.S.; Chen, Y.T.; Kuo, Y.L.; Chen, W.L.; Lee, M.S.; Wang, W. Mosaic tetrasomy 9p at amniocentesis: Prenatal diagnosis, molecular cytogenetic characterization, and literature review. Taiwan. J. Obstet. Gynecol. 2014, 53, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xie, L.S.; Wang, Y.; Mei, J. Prenatal diagnosis of mosaic tetrasomy 9p in a fetus with isolated persistent left superior vena cava. Taiwan. J. Obstet. Gynecol. 2015, 54, 204–205. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.L.; Glessner, J.T.; Porcu, E.; Niestroj, L.-M.; Ulirsch, J.; Kellaris, G.; Howrigan, D.P.; Everett, S.; Mohajeri, K.; Nuttle, X.; et al. A cross-disorder dosage sensitivity map of the human genome. MedRxiv 2021. [Google Scholar] [CrossRef]
- Van Opstal, D.; Srebniak, M.I. Cytogenetic confirmation of a positive NIPT result: Evidence-based choice between chorionic villus sampling and amniocentesis depending on chromosome aberration. Expert Rev. Mol. Diagn. 2016, 16, 513–520. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abnormalities (Occurrence in Published Cases) | Case 1 | Case 2 | References |
|---|---|---|---|
| CNS anomalies (n = 12; 55%) | − | + | [1,5,7,12,14,15,16,17,18,19,20,21] |
| Skeletal/limb anomalies (n = 12; 55%) | − | − | [2,3,5,7,12,13,16,19,20,21,22,23] |
| Cleft lip/palate (n = 10; 45%) | − | + | [1,3,5,12,16,19,21,23] |
| IUGR (n = 10; 45%) | − | − | [1,7,12,15,17,21,22,23,24] |
| Facial dysmorphism (n = 8; 36%) | − | + | [1,3,5,7,20,21,23] |
| Genito-urinary or renal anomalies (n = 7; 32%) | − | − | [1,3,5,15,16,18,21] |
| Amniotic fluid volume anomalies (n = 6; 27%) | − | − | [7,15,20,21,22] |
| Cardiac anomalies (n = 5; 23%) | − | + | [12,16,20,21,25] |
| Increased nuchal translucency (n = 4; 18%) | + | − | [1,3,5,21,23] |
| Absent nasal bone (n = 2; 9%) | − | + | [1,13] |
| Hydrops fetalis (n = 1; 5%) | − | + | [6] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaghi, M.; Janssens, K.; Hectors, W.; Loquet, P.; Blaumeiser, B. Tetrasomy 9p, a Prenatal Challenge: Two Novel Cases. Reprod. Med. 2022, 3, 42-49. https://doi.org/10.3390/reprodmed3010005
Zaghi M, Janssens K, Hectors W, Loquet P, Blaumeiser B. Tetrasomy 9p, a Prenatal Challenge: Two Novel Cases. Reproductive Medicine. 2022; 3(1):42-49. https://doi.org/10.3390/reprodmed3010005
Chicago/Turabian StyleZaghi, Mahtab, Katrien Janssens, Wim Hectors, Philip Loquet, and Bettina Blaumeiser. 2022. "Tetrasomy 9p, a Prenatal Challenge: Two Novel Cases" Reproductive Medicine 3, no. 1: 42-49. https://doi.org/10.3390/reprodmed3010005
APA StyleZaghi, M., Janssens, K., Hectors, W., Loquet, P., & Blaumeiser, B. (2022). Tetrasomy 9p, a Prenatal Challenge: Two Novel Cases. Reproductive Medicine, 3(1), 42-49. https://doi.org/10.3390/reprodmed3010005