
Synthesis of Amino-Acid-Based Nitroalkenes

 ,
, 
Abstract
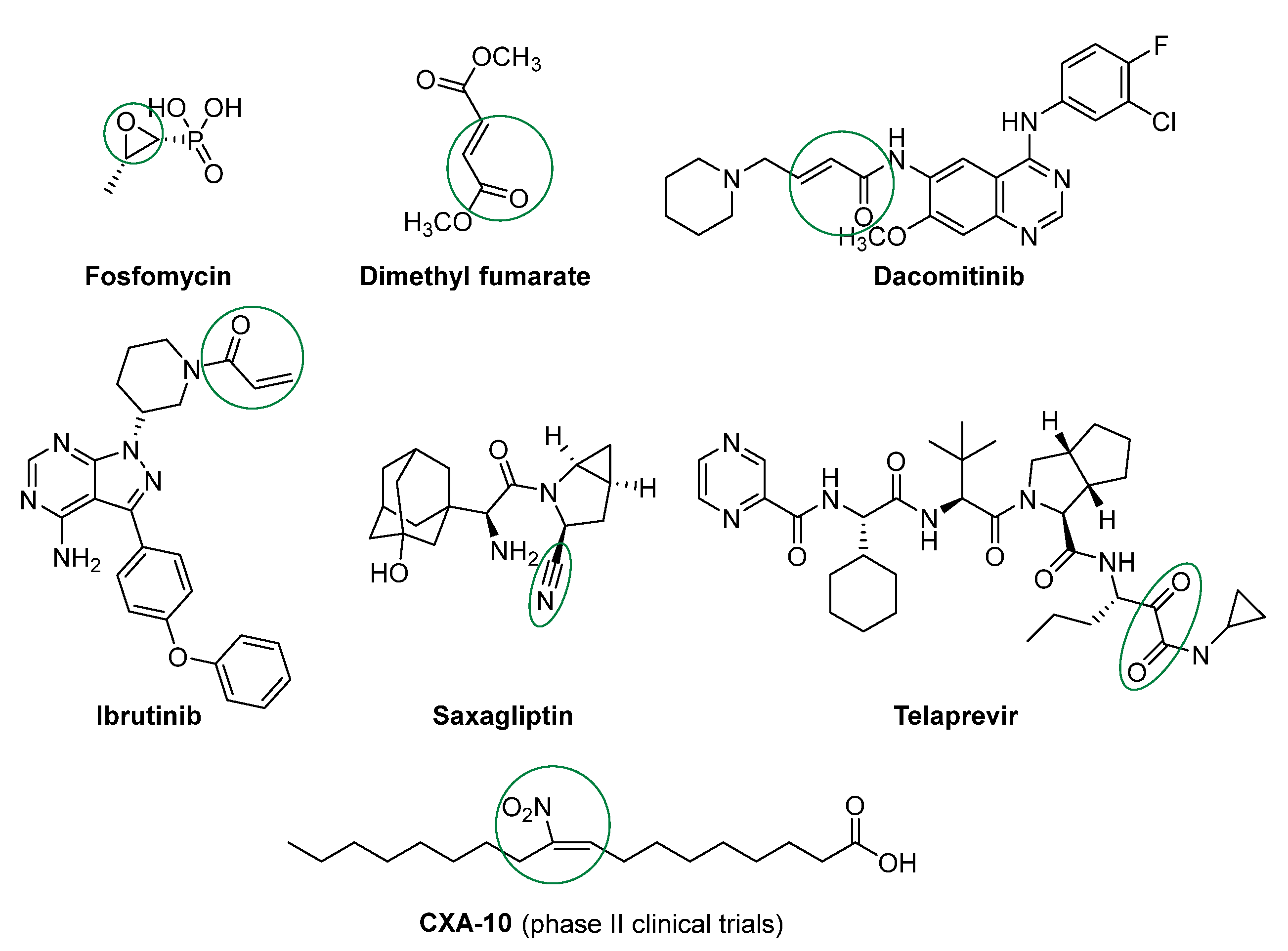
:1. Introduction
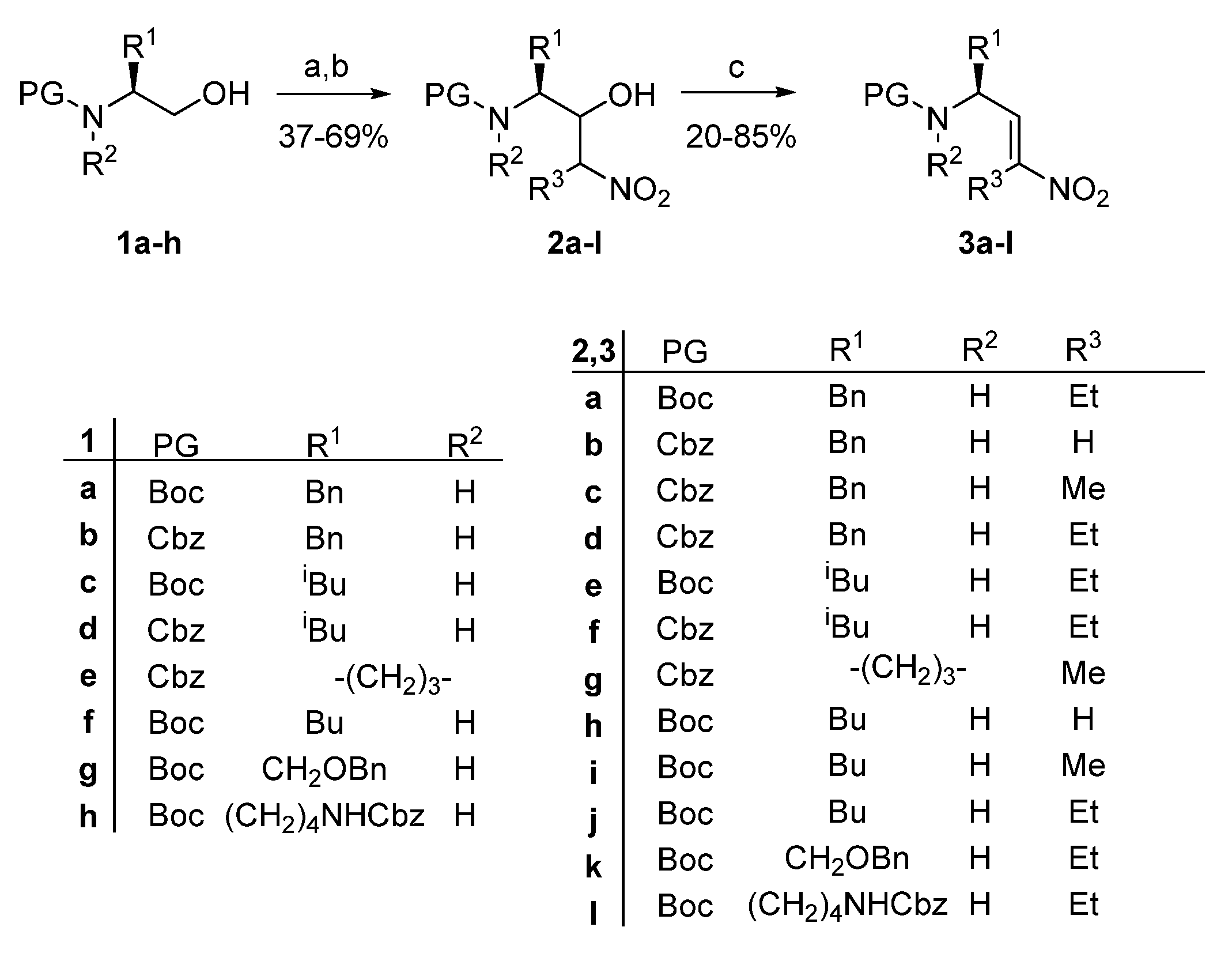
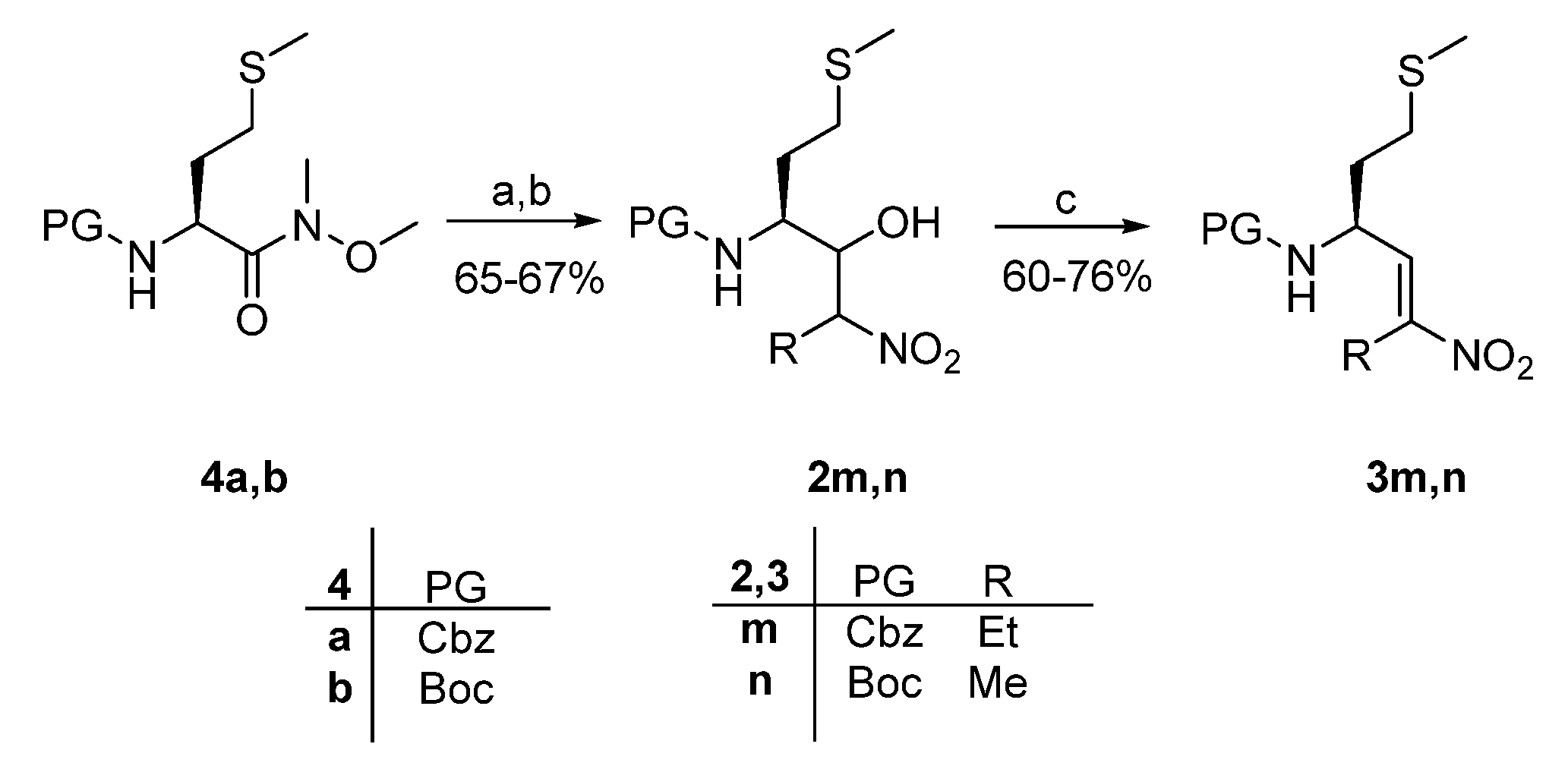
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. General Synthetic Procedures
4.2.1. General Method for the Oxidation of N-Protected 2-Amino Alcohols 1a-h to Aldehydes
4.2.2. General Methods for the Reduction of Weinreb Amides 4a,b to Aldehydes
4.2.3. General Methods for the Henry Reaction
4.2.4. General Methods for the Synthesis of Nitroalkenes 3a-n
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Samanta, I.; Mondal, A.; Liu, W.R. Covalent inhibition in drug discovery. ChemMedChem 2019, 14, 889–906. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.S.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising covalent warhead reactivity. Bioorg. Med. Chem. 2019, 27, 2066–2074. [Google Scholar] [CrossRef]
- Dalton, S.E.; Campos, S. Covalent small molecules as enabling platforms for drug discovery. ChemBioChem 2020, 21, 1080–1100. [Google Scholar] [CrossRef]
- Garner, R.M.; Mould, D.R.; Chieffo, C.; Jorkasky, D.K. Pharmacokinetic and pharmacodynamic effects of oral CXA-10, a nitro fatty acid, after single and multiple ascending doses in healthy and obese subjects. Clin. Transl. Sci. 2019, 12, 667–676. [Google Scholar] [CrossRef]
- Koutoulogenis, G.S.; Kokotos, G. Nitro Fatty Acids (NO2-FAs): An emerging class of bioactive fatty acids. Molecules 2021, 26, 7536. [Google Scholar] [CrossRef]
- Batthyany, C.; Schopfer, F.J.; Baker, P.R.S.; Durán, R.; Baker, L.M.S.; Huang, Y.; Cervenansky, C.; Branchaud, B.P.; Freeman, B.A. reversible post-translational modification of proteins by nitrated fatty acids in vivo. J. Biol. Chem. 2006, 281, 20450–20463. [Google Scholar] [CrossRef] [Green Version]
- Baker, L.M.S.; Baker, P.R.S.; Golin-Bisello, F.; Schopfer, F.J.; Fink, M.; Woodcock, S.R.; Branchaud, B.P.; Radi, R.; Freeman, B.A. Nitro-fatty acid reaction with glutathione and cysteine. Kinetic analysis of thiol alkylation by a Michael addition reaction. J. Biol. Chem. 2007, 282, 31085–31093. [Google Scholar] [CrossRef] [Green Version]
- Turell, L.; Vitturi, D.A.; Coitiño, E.L.; Lebrato, L.; Möller, M.N.; Sagasti, C.; Salvatore, S.R.; Woodcock, S.R.; Alvarez, B.; Schopfer, F.J. The chemical basis of thiol addition to nitro-conjugated linoleic acid, a protective cell-signaling lipid. J. Biol. Chem. 2017, 292, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Fazzari, M.; Vitturi, D.A.; Woodcock, S.R.; Salvatore, S.R.; Freeman, B.A.; Schopfer, F.J. Electrophilic fatty acid nitroalkenes are systemically transported and distributed upon esterification to complex lipids. J. Lipid Res. 2019, 60, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Khoo, N.K.H.; Li, L.; Salvatore, S.R.; Schopfer, F.J.; Freeman, B.A. Electrophilic fatty acid nitroalkenes regulate Nrf2 and NF-ΚB signaling: A medicinal chemistry investigation of structure-function relationships. Sci. Rep. 2018, 8, 2295. [Google Scholar] [CrossRef]
- Blanco, F.; Ferreira, A.M.; López, G.V.; Bonilla, L.; Gonzalez, M.; Cerecetto, H.; Trostchansky, A.; Rubbo, H. 6-Methylnitroarachidonate: A novel esterified nitroalkene that potently inhibits platelet aggregation and exerts cGMP-mediated vascular relaxation. Free Rad. Biol. Med. 2011, 50, 411–418. [Google Scholar] [CrossRef]
- Rodriguez-Duarte, J.; Galliussi, G.; Dapueto, R.; Rossello, J.; Malacrida, L.; Kamaid, A.; Schopfer, F.J.; Escande, C.; López, G.V.; Batthyany, C. A novel nitroalkene-α-tocopherol analogue inhibits inflammation and ameliorates atherosclerosis in Apo E knockout mice. Br. J. Pharmacol. 2019, 176, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Dapueto, R.; Rodriguez-Duarte, J.; Galliussi, G.; Kamaid, A.; Bresque, M.; Batthyany, C.; López, G.V.; Escande, C. A novel nitroalkene vitamin E analogue inhibits the NLRP3 inflammasome and protects against inflammation and glucose intolerance triggered by obesity. Redox Biol. 2021, 39, 101833. [Google Scholar] [CrossRef]
- Latorre, A.; Schirmeister, T.; Kesselring, J.; Jung, S.; Johé, P.; Hellmich, U.A.; Heilos, A.; Engels, B.; Krauth-Siegel, R.L.; Dirdjaja, N.; et al. Dipeptidyl nitroalkenes as potent reversible inhibitors of cysteine proteases rhodesain and cruzain. ACS Med. Chem. Lett. 2016, 7, 1073–1076. [Google Scholar] [CrossRef] [Green Version]
- Lecaille, F.; Kaleta, J.; Brömme, D. Human and parasitic papain-like cysteine proteases: Their role in physiology and pathology and recent developments in inhibitor design. Chem. Rev. 2002, 102, 4459–4488. [Google Scholar] [CrossRef]
- Arafet, K.; González, F.V.; Moliner, V. Quantum Mechanics/Molecular Mechanics studies of the mechanism of cysteine proteases inhibition by dipeptidyl nitroalkenes. Chem. A Eur. J. 2020, 26, 2002–2012. [Google Scholar] [CrossRef]
- Arafet, K.; Serrano Aparicio, N.; Lodola, A.; Mulholland, A.J.; González, F.V.; Świderek, K.; Moliner, V. Mechanism of inhibition of SARS-CoV-2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 2021, 12, 1433–1444. [Google Scholar] [CrossRef]
- Hassan, M.; Nde, C.N.; Manolikakes, G. Synthesis of nitroolefins via the direct nitration of alkenes. SynOpen 2021, 5, 229–231. [Google Scholar]
- Fioravanti, S.; Pellacani, L.; Tardella, P.A.; Vergari, M.C. Facile and highly stereoselective one-pot synthesis of either (E)- or (Z)-nitro alkenes. Org. Lett. 2008, 10, 1449–1451. [Google Scholar] [CrossRef]
- Woodcock, S.R.; Marwitz, A.J.V.; Bruno, P.; Branchaud, B.P. Synthesis of nitrolipids. All four possible diastereomers of nitrooleic acids: (E)- and (Z)-, 9- and 10-nitro-octadec-9-enoic acids. Org. Lett. 2006, 8, 3931–3934. [Google Scholar] [CrossRef]
- Gorczynski, M.J.; Smitherman, P.K.; Akiyama, T.E.; Wood, H.B.; Berger, J.P.; King, S.B.; Morrow, C.S. Activation of Peroxisome Proliferator-Activated Receptor γ (PPARγ) by nitroalkene fatty acids: Importance of nitration position and degree of unsaturation. J. Med. Chem. 2009, 52, 4631–4639. [Google Scholar] [CrossRef] [Green Version]
- Hock, K.J.; Grimmer, J.; Göbel, D.; Gasaya, G.G.T.; Roos, J.; Maucher, I.V.; Kühn, B.; Fettel, J.; Maier, T.J.; Manolikakes, G. Modular regiospecific synthesis of nitrated fatty acids. Synthesis 2016, 49, 615–636. [Google Scholar]
- Hassan, M.; Krieg, S.C.; Nde, C.N.; Roos, J.; Maier, T.J.; El Rady, E.A.; Raslan, M.A.; Sadek, K.U.; Manolikakes, G. Streamlined one-pot synthesis of nitro fatty acids. Eur. J. Org. Chem. 2021, 15, 2239–2252. [Google Scholar] [CrossRef]
- Bhat, C.; Tilve, S.G. Synthesis of (-)-hygrine, (-)-norhygrine, (-)-pseudohygroline and (-)-hygroline via Nef reaction. Tetrahedron Lett. 2011, 52, 6566–6568. [Google Scholar] [CrossRef]
- Bhat, C.; Tilve, S.G. Henry-Nef reaction: A practical and versatile chiral pool route to 2-substituted pyrrolidine and piperidine alkaloids. Tetrahedron 2013, 69, 6129–6143. [Google Scholar] [CrossRef]
- Hanessian, S.; Devasthale, P.V. Generation of functional diversity via nitroaldol condensations of α-aminoacid aldehydes-A new and stereocontrolled route to acyclic 1,3-diamino-2-alcohols. Tetrahedron Lett. 1996, 37, 987–990. [Google Scholar] [CrossRef]
- Pereira, V.L.P.; Moura, A.L.; Vieira, D.P.; de Carvalho, L.L.; Torres, E.R.; da Costa, J.S. A versatile and efficient approach for the synthesis of chiral 1,3-nitroamines and 1,3-diamines via conjugate addition to new (S,E)-γ-aminated nitroalkenes derived from L-α-amino acids. Beilstein J. Org. Chem. 2013, 9, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Meirelis, F.P.; Vieira, B.G.N.; Pereira, V.L.P. Divergent and diastereoselective synthesis of α-monosubstituted and trans-α,β-disubstituted γ-lactams from (S)-N,N-dibenzyl-α-amino aldehydes via Henry and Michael reactions. Synthesis 2020, 52, 3650–3656. [Google Scholar]
- Ambroise, L.; Jackson, R.F.W. Stereoselective synthesis of anti-β-amino-α-hydroxy acid derivatives using nucleophilic epoxidation of 1-arylthio-l-nitroalkenes. Tetrahedron Lett. 1996, 37, 2311–2314. [Google Scholar] [CrossRef]
- Kokotos, G. A convenient one-pot conversion of N-protected amino acids and peptides into alcohols. Synthesis 1990, 1990, 299–301. [Google Scholar] [CrossRef]
- Kokotos, G.; Noula, C. Selective one-pot conversion of carboxylic acids into alcohols. J. Org. Chem. 1996, 61, 6994–6996. [Google Scholar] [CrossRef] [PubMed]
- Loukas, V.; Noula, C. Efficient protocols for the synthesis of enantiopure γ-amino acids with proteinogenic side chains. J. Peptide Sci. 2003, 9, 312–319. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Entry | Aldehyde | Method | Nitroalkane | Product | Isolated Yield (%) |
|---|---|---|---|---|---|
| 1 |  | A | 1-Nitropropane | 2a | 37 |
| 2 |  | A | Nitromethane | 2b | 69 |
| 3 |  | A | Nitroethane | 2c | 40 |
| 4 |  | A | 1-Nitropropane | 2d | 57 |
| 5 |  | A | 1-Nitropropane | 2e | 51 |
| 6 |  | B | 1-Nitropropane | 2f | 42 |
| 7 |  | B | Nitroethane | 2g | - b |
| 8 |  | A | Nitromethane | 2h | 58 |
| 9 |  | A | Nitroethane | 2i | 46 |
| 10 |  | A | 1-Nitropropane | 2j | 40 |
| 11 |  | A or B | 1-Nitropropane | 2k | 36 or 42 |
| 12 |  | A | 1-Nitropropane | 2l | 36 |
| 13 |  | A | Nitroethane | 2m | 67 |
| 14 |  | A | 1-Nitropropane | 2n | 65 |
| Entry | Nitro Alcohol | Method | Nitroalkene | Reaction Time (h) | Isolated Yield (%) |
|---|---|---|---|---|---|
| 1 | 2a | C |  | 72 | 47 |
| 2 | 2b | D |  | 1 | 42 |
| 3 | 2c | E |  | 2 | 42 |
| 4 | 2d | C |  | 72 | 53 |
| 5 | 2e | C |  | 72 | 58 |
| 6 | 2f | E |  | 7 | 41 |
| 7 | 2g | E |  | 5 | 20 a |
| 8 | 2h | D |  | 1 | 82 |
| 9 | 2i | E |  | 2 | 85 |
| 10 | 2j | C |  | 72 | 72 |
| 11 | 2k | C or E |  | 72 | 35 or 35 |
| 12 | 2l | C |  | 48 | 51 |
| 13 | 2m | E |  | 24 | 60 |
| 14 | 2n | E |  | 1.5 | 76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerogianni, V.-E.; Koutoulogenis, G.S.; Gerokonstantis, D.T.; Kokotos, G. Synthesis of Amino-Acid-Based Nitroalkenes. Organics 2022, 3, 137-149. https://doi.org/10.3390/org3020011
Gerogianni V-E, Koutoulogenis GS, Gerokonstantis DT, Kokotos G. Synthesis of Amino-Acid-Based Nitroalkenes. Organics. 2022; 3(2):137-149. https://doi.org/10.3390/org3020011
Chicago/Turabian StyleGerogianni, Velisaria-Eleni, Giorgos S. Koutoulogenis, Dimitrios Triantafyllos Gerokonstantis, and George Kokotos. 2022. "Synthesis of Amino-Acid-Based Nitroalkenes" Organics 3, no. 2: 137-149. https://doi.org/10.3390/org3020011
APA StyleGerogianni, V. -E., Koutoulogenis, G. S., Gerokonstantis, D. T., & Kokotos, G. (2022). Synthesis of Amino-Acid-Based Nitroalkenes. Organics, 3(2), 137-149. https://doi.org/10.3390/org3020011