

Long-Term Survival of a Child with Atypical Teratoid-Rhabdoid Tumor and Acute Lymphoblastic Leukemia: A Case Report


 and
and 
Abstract
:1. Introduction
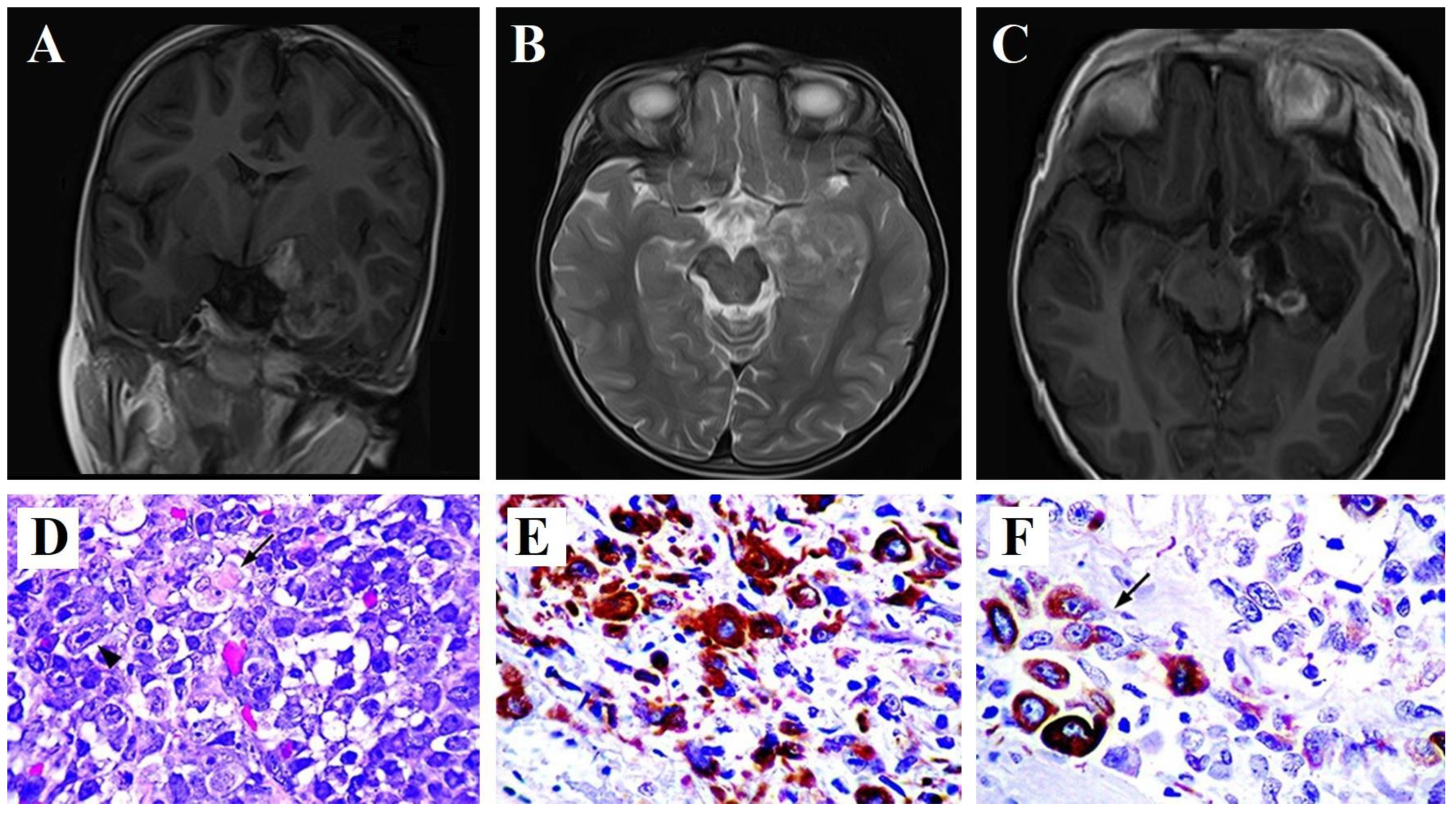
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhattacharjee, M.; Hicks, J.; Langford, L.; Dauser, R.; Strother, D.; Chintagumpala, M.; Horowitz, M.; Cooley, L.; Vogel, H. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. Ultrastruct. Pathol. 1997, 21, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Judkins, A.R.; Eberhart, C.G.; Wesseling, P. Atypical teratoid/rhabdoid tumour. In WHO Classification of Tumours of the Central Nervous System, 4th ed.; Louis, D.N., Ohgaki, H., Wiestler, O.D., Cavenee, W.K., Eds.; International Agency for Research on Cancer: Lyon, France, 2007; Volume 1, pp. 147–149. [Google Scholar]
- Grundy, R.; Wilne, S.; Robinson, K.; Ironside, J.; Cox, T.; Chong, W.; Michalski, A.; Campbell, R.; Bailey, C.; Thorp, N.; et al. Primary postoperative chemotherapy without radiotherapy for treatment of brain tumours other than ependymoma in children under 3 years: Results of the first UKCCSG/SIOP CNS 9204 trial. Eur. J. Cancer 2010, 46, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, J.B.; Palmer, N.F. Histopathology and prognosis of Wilms tumors: Results from the First National Wilms’ Tumor Study. Cancer 1978, 41, 1937–1948. [Google Scholar] [CrossRef] [PubMed]
- Frühwald, M.C.; Biegel, J.A.; Bourdeaut, F.; Roberts, C.W.; Chi, S.N. Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol. 2016, 18, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.; Johann, P.D.; Grabovska, Y.; Andrianteranagna, M.J.D.D.; Yao, F.; Frühwald, M.; Hasselblatt, M.; Bourdeaut, F.; Williamson, D.; Huang, A.; et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol. 2020, 22, 613–624, Erratum in Neuro Oncol. 2022, 24, 1213. [Google Scholar] [CrossRef] [PubMed]
- Torchia, J.; Golbourn, B.; Feng, S.; Ho, K.C.; Sin-Chan, P.; Vasiljevic, A.; Norman, J.D.; Guilhamon, P.; Garzia, L.; Agamez, N.R.; et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell 2016, 30, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Nowak, J.; Nemes, K.; Hohm, A.; Vandergrift, L.A.; Hasselblatt, M.; Johann, P.D.; Kool, M.; Frühwald, M.C.; Warmuth-Metz, M. Magnetic resonance imaging surrogates of molecular subgroups in atypical teratoid/rhabdoid tumor. Neuro Oncol. 2018, 20, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.M.; Hamideh, D.; Bahrami, A.; Orr, B.A.; Lin, T.; Pounds, S.; Zambetti, G.P.; Pappo, A.S.; Gajjar, A.; Agnihotri, S.; et al. Malignant rhabdoid tumors originating within and outside the central nervous system are clinically and molecularly heterogeneous. Acta Neuropathol. 2018, 136, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.E.; Johann, P.D.; Milne, K.; Zapatka, M.; Buellesbach, A.; Ishaque, N.; Iskar, M.; Erkek, S.; Wei, L.; Tessier-Cloutier, B.; et al. Identification and analyses of extra-cranial and cranial rhabdoid tumor molecular subgroups reveal tumors with cytotoxic T cell infiltration. Cell Rep. 2019, 29, 2338–2354.e2337. [Google Scholar] [CrossRef]
- Oberlick, E.M.; Rees, M.G.; Seashore-Ludlow, B.; Vazquez, F.; Nelson, G.M.; Dharia, N.V.; Weir, B.A.; Tsherniak, A.; Ghandi, M.; Krill-Burger, J.M.; et al. Small-molecule and CRISPR screening converge to reveal receptor tyrosine kinase dependencies in pediatric rhabdoid tumors. Cell Rep. 2019, 28, 2331–2344.e2338. [Google Scholar] [CrossRef]
- Lakhdar, F.; Benzagmout, M.; Arkha, Y.; Chakour, K.; Chaoui, M.E.F. ATRT of lateral ventricle in a child: A Rare Tumor at a Very Rare Location. Asian J. Neurosurg. 2020, 15, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Lafay-Cousin, L.; Hawkins, C.; Carret, A.; Johnston, D.; Zelcer, S.; Wilson, B.; Jabado, N.; Scheinemann, K.; Eisenstat, D.; Fryer, C.; et al. Central nervous system atypical teratoid rhabdoid tumours: The Canadian Paediatric Brain Tumour Consortium experience. Eur. J. Cancer 2012, 48, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Vejmelkova, K.; Pokorna, P.; Noskova, K.; Faustmannova, A.; Drabova, K.; Pavelka, Z.; Bajciova, V.; Broz, M.; Tinka, P.; Jezova, M.; et al. Tazemetostat in the therapy of pediatric INI1-negative malignant rhabdoid tumors. Sci. Rep. 2023, 13, 21623. [Google Scholar] [CrossRef] [PubMed]
- Oeffinger, K.C.; Mertens, A.C.; Sklar, C.A.; Kawashima, T.; Hudson, M.M.; Meadows, A.T.; Friedman, D.L.; Marina, N.; Hobbie, W.; Kadan-Lottick, N.S.; et al. Chronic health conditions in adult survivors of childhood cancer. N. Engl. J. Med. 2006, 355, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.T.; Liu, Q.; Yasui, Y.; Neglia, J.P.; Leisenring, W.; Robison, L.L.; Mertens, A.C. Late mortality among 5-year survivors of childhood cancer: A summary from the Childhood Cancer Survivor Study. J. Clin. Oncol. 2009, 27, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, H. Molecular targeted therapies for pediatric atypical teratoid/rhabdoid tumors. Pediatr. Investig. 2022, 6, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Kuge, A.; Sato, S.; Sakurada, K.; Takemura, S.; Kayama, T. Atypical teratoid rhabdoid tumor located in the pineal region following prophylactic irradiation for acute lymphoblastic leukemia. Brain Tumor Pathol. 2012, 29, 177–181. [Google Scholar] [CrossRef]
- MacDonald, T.J.; Rood, B.R.; Santi, M.R.; Vezina, G.; Bingaman, K.; Cogen, P.H.; Packer, R.J. Advances in the diagnosis, molecular genetics, and treatment of pediatric embryonal CNS tumors. Oncologist 2003, 8, 174–186. [Google Scholar] [CrossRef]
- Burger, P.C.; Yu, I.T.; Tihan, T.; Friedman, H.S.; Strother, D.R.; Kepner, J.L.; Duffner, P.K.; Kun, L.E.; Perlman, E.J. Atypical teratoid/rhabdoid tumor of the central nervous system: A highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: A Pediatric Oncology Group study. Am. J. Surg. Pathol. 1998, 22, 1083–1092. [Google Scholar] [CrossRef]
- Rorke, L.B.; Packer, R.J.; Biegel, J.A. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: Definition of an entity. J. Neurosurg. 1996, 85, 56–65. [Google Scholar] [CrossRef]
- Kumirova, E.V.; Ozerov, S.S.; Ryzhova, M.V.; Konovalov, D.M.; Shekhtman, A.P.; Emtsova, V.V.; Vyazova, Y.V.; Andrianov, M.M.; Abbasova, E.V.; Gvozdev, A.A.; et al. A rare embryonic tumor of the central nervous system–neuroblastoma with FOXR2 activation FOXR. Russ. J. Pediatr. Hematol. Oncol. 2022, 9, 11–21. [Google Scholar] [CrossRef]
- Montemurro, N.; Pahwa, B.; Tayal, A.; Shukla, A.; De Jesus Encarnacion, M.; Ramirez, I.; Nurmukhametov, R.; Chavda, V.; De Carlo, A. Macrophages in Recurrent Glioblastoma as a Prognostic Factor in the Synergistic System of the Tumor Microenvironment. Neurol. Int. 2023, 15, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Zheludkova, O.G.; Korshunov, A.G.; Gorbatykh, S.V.; Livshitz, M.I.; Popov, V.E.; Tarasova, I.S.; Gorelyshex, S.K.; Ozerova, V.I.; Shcherbenko, O.I.; Litvinov, D.V.; et al. Malignant teratoid-rabdoid tumors of the central nervous system in children. Quest. Hematol. Oncol. Immunopathol. Pediatr. 2003, 3, 32–39. [Google Scholar]
- D’arco, F.; Khan, F.; Mankad, K.; Ganau, M.; Caro-Dominguez, P.; Bisdas, S. Differential diagnosis of posterior fossa tumours in children: New insights. Pediatr. Radiol. 2018, 48, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Beylerli, O.; Ramirez, M.d.J.E.; Shumadalova, A.; Ilyasova, T.; Zemlyanskiy, M.; Beilerli, A.; Montemurro, N. Cell-Free miRNAs as Non-Invasive Biomarkers in Brain Tumors. Diagnostics 2023, 13, 2888. [Google Scholar] [CrossRef] [PubMed]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.; Sturm, D.; Hermann, C.; Wang, M.S.; et al. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Dinikina, Y.V.; Belogurova, M.B. Atypical teratoid/rhabdoid tumors of the central nervous system in children: The state of the problem today. Literature review. RŽDGiO 2018, 4, 150. [Google Scholar] [CrossRef]
- Frühwald, M.C.; Hasselblatt, M.; Nemes, K.; Bens, S.; Steinbügl, M.; Johann, P.D.; Kerl, K.; Hauser, P.; Quiroga, E.; Solano-Paez, P.; et al. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol. 2020, 22, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.T.; Strother, D.R.; Judkins, A.R.; Burger, P.C.; Pollack, I.F.; Krailo, M.D.; Buxton, A.B.; Williams-Hughes, C.; Fouladi, M.; Mahajan, A.; et al. Efficacy of high-dose chemotherapy and three-dimensional conformal radiation for atypical teratoid/rhabdoid tumor: A report from the children’s oncology group trial ACNS0333. J. Clin. Oncol. 2020, 38, 1175–1185. [Google Scholar] [CrossRef]
- Federico, A.; Thomas, C.; Miskiewicz, K.; Woltering, N.; Zin, F.; Nemes, K.; Bison, B.; Johann, P.D.; Hawes, D.; Bens, S.; et al. ATRT–SHH comprises three molecular subgroups with characteristic clinical and histopathological features and prognostic significance. Acta Neuropathol. 2022, 143, 697–711. [Google Scholar] [CrossRef]
- Turcotte, L.M.; Liu, Q.; Yasui, Y.; Arnold, M.A.; Hammond, S.; Howell, R.M.; Smith, S.A.; Weathers, R.E.; Henderson, T.O.; Gibson, T.M.; et al. Temporal Trends in Treatment and Subsequent Neoplasm Risk Among 5-Year Survivors of Childhood Cancer, 1970–2015. J. Am. Med. Assoc. 2017, 317, 814–824. [Google Scholar] [CrossRef] [PubMed]
- United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). UNSCEAR 2006 Report. In Annex A. Epidemiological Studies of Radiation and Cancer; United Nations: New York, NY, USA, 2008; Volume 1, p. 1. [Google Scholar]
- Armstrong, B.; Brenner, D.J.; Baverstock, K.; Cardis, E.; Baguley, B.C. A Review of Human Carcinogens; International Agency for Research on Cancer: Lyon, France, 2012; Volume 1, p. 25. [Google Scholar]
- Allodji, R.S.; Tucker, M.A.; Hawkins, M.M.; Le Deley, M.; Veres, C.; Weathers, R.; Howell, R.; Winter, D.; Haddy, N.; Rubino, C.; et al. Role of radiotherapy and chemotherapy in the risk of leukemia after childhood cancer: An international pooled analysis. Int. J. Cancer 2021, 148, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
- Stary, J.; Zimmermann, M.; Campbell, M.; Castillo, L.; Dibar, E.; Donska, S.; Gonzalez, A.; Izraeli, S.; Janic, D.; Jazbec, J.; et al. Intensive chemotherapy for childhood acute lymphoblastic leukemia: Results of the randomized intercontinental trial ALL IC-BFM 2002. J. Clin. Oncol. 2014, 32, 174–184. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreevna, K.M.; Vyacheslavovna, K.E.; Valeryevna, G.S.; Gunai Nariman, M.; Igorevich, L.M.; Yegorovich, C.G.; Nikolaevich, K.A.; Nikolaevich, U.V.; Encarnacion Ramirez, M.d.J.; Montemurro, N. Long-Term Survival of a Child with Atypical Teratoid-Rhabdoid Tumor and Acute Lymphoblastic Leukemia: A Case Report. Surgeries 2024, 5, 184-193. https://doi.org/10.3390/surgeries5020018
Andreevna KM, Vyacheslavovna KE, Valeryevna GS, Gunai Nariman M, Igorevich LM, Yegorovich CG, Nikolaevich KA, Nikolaevich UV, Encarnacion Ramirez MdJ, Montemurro N. Long-Term Survival of a Child with Atypical Teratoid-Rhabdoid Tumor and Acute Lymphoblastic Leukemia: A Case Report. Surgeries. 2024; 5(2):184-193. https://doi.org/10.3390/surgeries5020018
Chicago/Turabian StyleAndreevna, Kolcheva Maria, Kumirova Ella Vyacheslavovna, Gorbatykh Svetlana Valeryevna, Makhmudova Gunai Nariman, Livshits Matvey Igorevich, Chmutin Gennadiy Yegorovich, Kislyakov Alexey Nikolaevich, Umerenkov Viktor Nikolaevich, Manuel de Jesus Encarnacion Ramirez, and Nicola Montemurro. 2024. "Long-Term Survival of a Child with Atypical Teratoid-Rhabdoid Tumor and Acute Lymphoblastic Leukemia: A Case Report" Surgeries 5, no. 2: 184-193. https://doi.org/10.3390/surgeries5020018
APA StyleAndreevna, K. M., Vyacheslavovna, K. E., Valeryevna, G. S., Gunai Nariman, M., Igorevich, L. M., Yegorovich, C. G., Nikolaevich, K. A., Nikolaevich, U. V., Encarnacion Ramirez, M. d. J., & Montemurro, N. (2024). Long-Term Survival of a Child with Atypical Teratoid-Rhabdoid Tumor and Acute Lymphoblastic Leukemia: A Case Report. Surgeries, 5(2), 184-193. https://doi.org/10.3390/surgeries5020018