
Synthesis of Porphyrins with ABAB Symmetry from Dipyrromethanes as Potential Phototherapeutic Agents †


Abstract
:1. Introduction
2. Materials and Methods
2.1. Equipment and Chemical Substances
2.2. Synthesis
2.3. Spectroscopic Studies
3. Results and Discussion
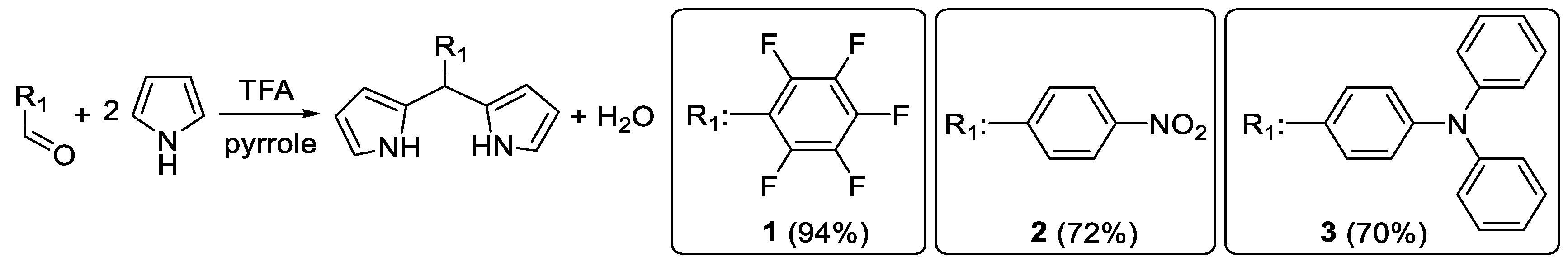
3.1. Synthesis of Dipyrromethanes
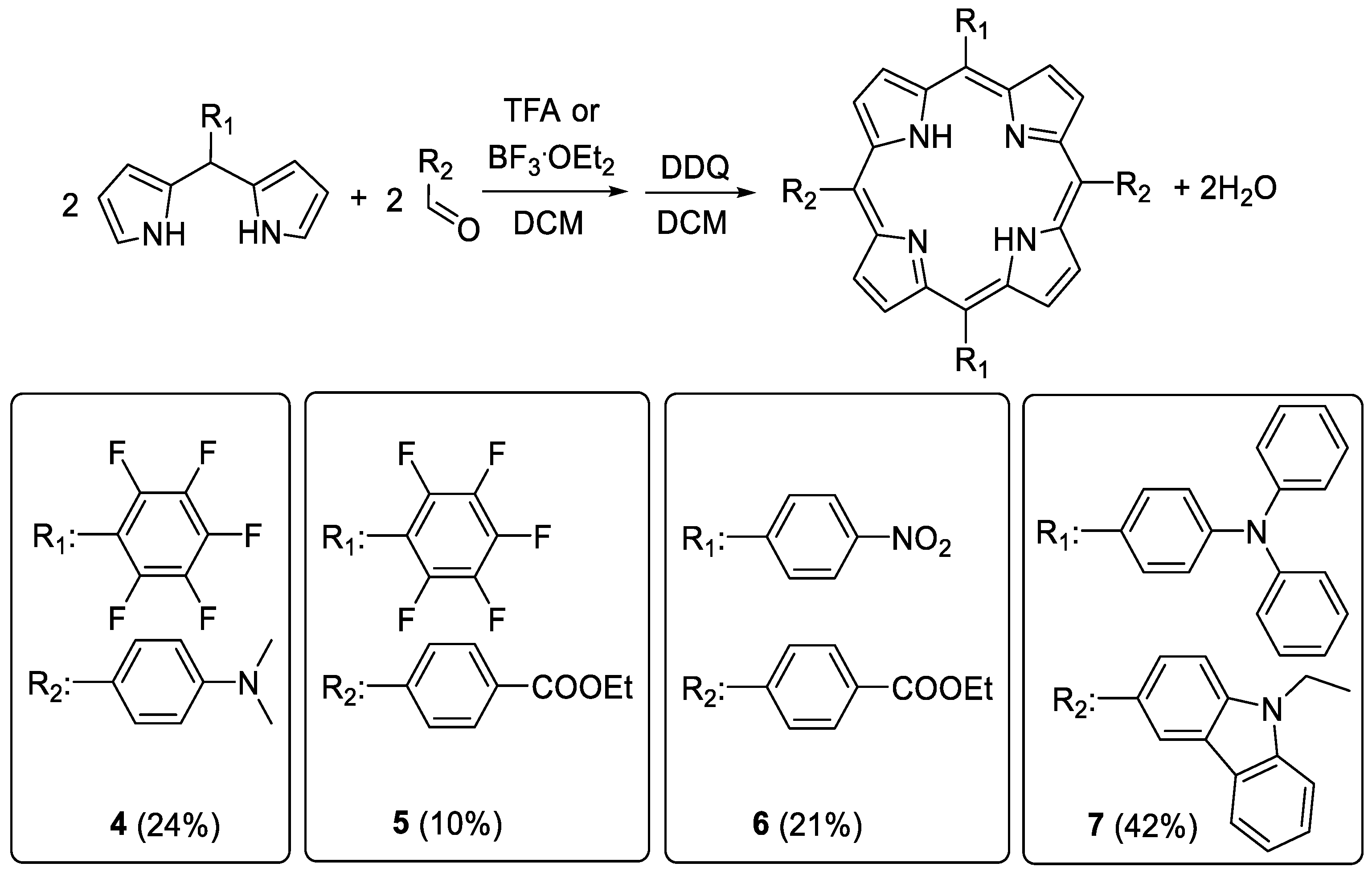
3.2. Synthesis of ABAB-Porphyrins
3.3. UV-Visible Absorption Spectroscopic Properties
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Martinez De Pinillos Bayona, A.; Mroz, P.; Thunshelle, C.; Hamblin, M.R. Design features for optimization of tetrapyrrole macrocycles as antimicrobial and anticancer photosensitizers. Chem. Biol. Drug. Des. 2017, 89, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Alves, E.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Cunha, Â.; Nadais, H.; Almeida, A. Potential applications of porphyrins in photodynamic inactivation beyond the medical scope. J. Photochem. Photobiol. C Photochem. Rev. 2015, 22, 34–57. [Google Scholar] [CrossRef]
- Heredia, D.A.; Martínez, S.R.; Durantini, A.M.; Pérez, M.E.; Mangione, M.I.; Durantini, J.E.; Gervaldo, M.A.; Otero, L.A.; Durantini, E.N. Antimicrobial photodynamic polymeric films bearing biscarbazol triphenylamine end-capped dendrimeric Zn(II) porphyrin. ACS Appl. Mater. Interfaces 2019, 11, 27574–27587. [Google Scholar] [CrossRef] [PubMed]
- Scanone, A.C.; Gsponer, N.S.; Alvarez, M.G.; Heredia, D.A.; Durantini, A.M.; Durantini, E.N. Magnetic nanoplatforms for in situ modification of macromolecules: Synthesis, characterization and photoinactivating power of cationic nanoiman-porphyrin conjugates. ACS Appl. Bio Mater. 2020, 3, 5930–5940. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, M.B.; Spesia, M.B.; Milanesio, M.E.; Durantini, E.N. Synthesis, spectroscopic properties and photodynamic activity of porphyrin-fullerene C60 dyads with application in the photodynamic inactivation of Staphylococcus aureus. Eur. J. Med. Chem. 2014, 83, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Scanone, A.C.; Gsponer, N.S.; Alvarez, M.G.; Durantini, E.N. Porphyrins containing basic aliphatic amino groups as potential broad-spectrum antimicrobial agents. Photodiagn. Photodyn. Ther. 2018, 24, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Durantini, E.N.; Silber, J.J. Synthesis of 5-(4-acetamidophenyl)-10,15,20-tris(4-substituted phenyl) porphyrins using dipyrromethanes. Synth. Commun. 1999, 29, 3353–3368. [Google Scholar] [CrossRef]
- Lindsey, J.S. Synthetic routes to meso-patterned porphyrins. Acc. Chem. Res. 2010, 43, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Lindsey, J.S. One-flask synthesis of meso-substituted dipyrromethanes and their application in the synthesis of trans-substituted porphyrin building blocks. Tetrahedron 1994, 50, 11427–11440. [Google Scholar] [CrossRef]
- Gutsche, C.S.; Ortwerth, M.; Gräfe, S.; Flanagan, K.J.; Senge, M.O.; Reissig, H.-U.; Kulak, N.; Wiehe, A. Nucleophilic aromatic substitution on pentafluorophenyl-substituted dipyrranes and tetrapyrroles as a route to multifunctionalized chromophores for potential application in photodynamic. Chem. Eur. J. 2016, 22, 13953–13964. [Google Scholar] [CrossRef] [PubMed]
- Megiatto, J.D.; Antoniuk-Pablant, A.; Sherman, B.D.; Kodis, G.; Gervaldo, M.; Moore, T.A.; Moore, A.L.; Gust, D. Mimicking the electron transfer chain in photosystem II with a molecular triad thermodynamically capable of water oxidation. Proc. Natl. Acad. Sci. USA 2012, 109, 15578–15583. [Google Scholar] [CrossRef] [PubMed]
- Durantini, J.; Morales, G.M.; Santo, M.; Funes, M.; Durantini, E.N.; Fungo, F.; Dittrich, T.; Otero, L.; Gervaldo, M. Synthesis and characterization of porphyrin electrochromic and photovoltaic electropolymers. Org. Electron. 2012, 13, 604–614. [Google Scholar] [CrossRef]
- Baskin, J.S.; Yu, H.Z.; Zewail, A.H. Ultrafast dynamics of porphyrins in the condensed phase: I. free base tetraphenylporphyrin. J. Phys. Chem. A 2002, 106, 9837–9844. [Google Scholar] [CrossRef]
- Mora, S.J.; Cormick, M.P.; Milanesio, M.E.; Durantini, N.E. The photodynamic activity of a novel porphyrin derivative bearing a fluconazole structure in different media and against Candida albicans. Dyes Pigm. 2010, 87, 234–240. [Google Scholar] [CrossRef]
- Kooriyaden, F.R.; Sujatha, S.; Arunkumar, C. Study of scrambling in porphyrin forming reactions: Synthesis, structure, photophysical, electrochemical and antimicrobial studies. Polyhedron 2017, 128, 85–94. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PS | Absorptionmax (nm) | εSoret a | Fluorescencemax (nm) | ΦF b |
|---|---|---|---|---|
| 4 | 418 510 542 589 647 | 4.67 × 105 | 651 712 | 0.063 ± 0.003 |
| 5 | 420 512 543 590 648 | 4.72 × 105 | 652 712 | 0.054 ± 0.002 |
| 6 | 421 516 551 591 647 | 4.72 × 105 | 652 717 | 0.10 ± 0.01 |
| 7 | 428 520 563 596 653 | 3.15 × 105 | 668 729 | 0.12 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boarini, M.B.; Pérez, M.E.; Milanesio, M.E.; Durantini, E.N. Synthesis of Porphyrins with ABAB Symmetry from Dipyrromethanes as Potential Phototherapeutic Agents. Chem. Proc. 2021, 3, 133. https://doi.org/10.3390/ecsoc-24-08305
Boarini MB, Pérez ME, Milanesio ME, Durantini EN. Synthesis of Porphyrins with ABAB Symmetry from Dipyrromethanes as Potential Phototherapeutic Agents. Chemistry Proceedings. 2021; 3(1):133. https://doi.org/10.3390/ecsoc-24-08305
Chicago/Turabian StyleBoarini, Milena B., María E. Pérez, María E. Milanesio, and Edgardo N. Durantini. 2021. "Synthesis of Porphyrins with ABAB Symmetry from Dipyrromethanes as Potential Phototherapeutic Agents" Chemistry Proceedings 3, no. 1: 133. https://doi.org/10.3390/ecsoc-24-08305
APA StyleBoarini, M. B., Pérez, M. E., Milanesio, M. E., & Durantini, E. N. (2021). Synthesis of Porphyrins with ABAB Symmetry from Dipyrromethanes as Potential Phototherapeutic Agents. Chemistry Proceedings, 3(1), 133. https://doi.org/10.3390/ecsoc-24-08305