Stress-Induced Alteration in Chloride Transporters in the Trigeminal Nerve May Explain the Comorbidity between Depression and Migraine

{kind=link}
Abstract
:1. Introduction
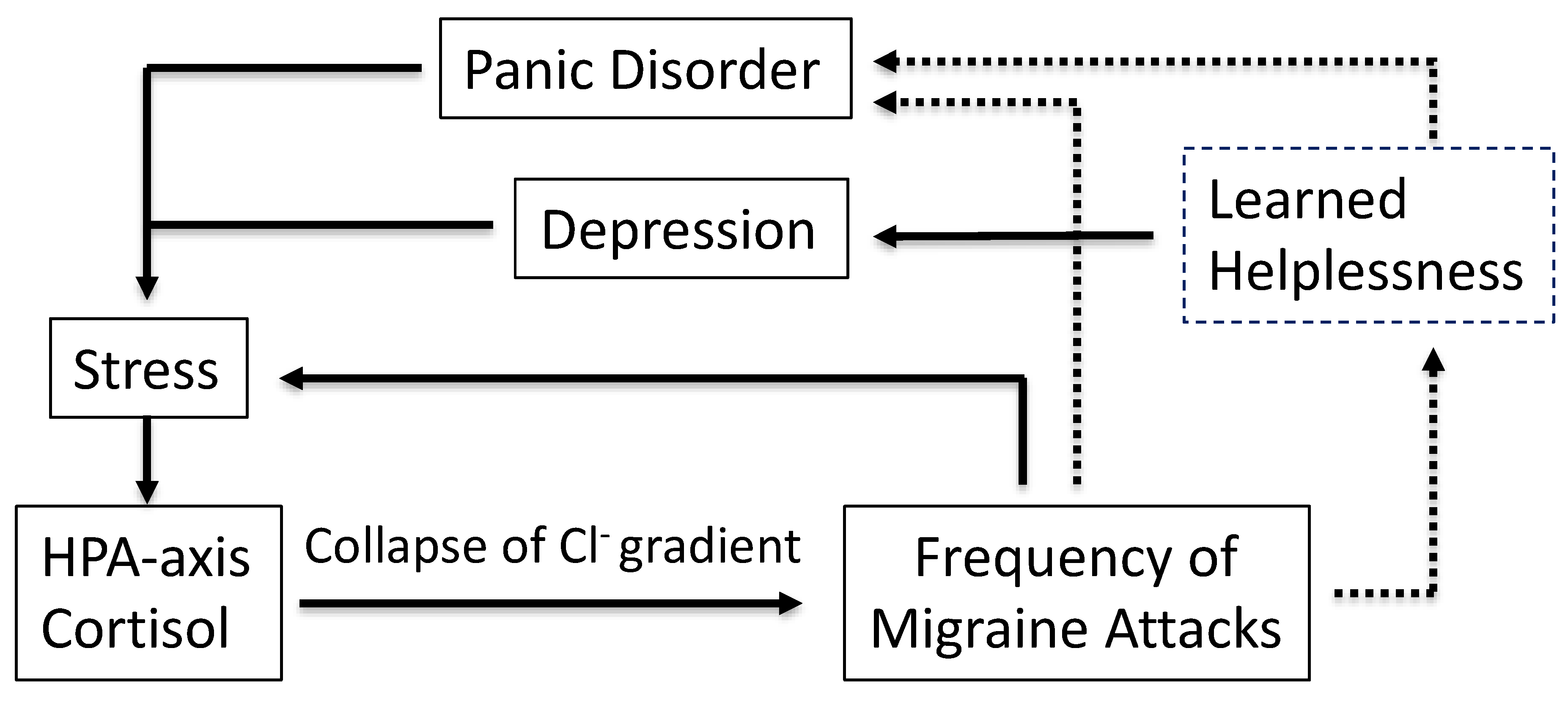
2. Migraine Is Co-Morbid with Panic Disorder and Depression
3. GABA Receptors and Functional Responses of the Trigeminal Nerve
4. Stress Modifies the Activity of Chloride Transporters
5. Discussion
6. Conclusions
Funding
Conflicts of Interest
References
- Headache Classification Committee of the International Headache Society. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013, 33, 629–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, R.; Jakubowski, M. Neural substrate of depression during migraine. Neurol. Sci. 2009, 30 (Suppl. 1), S27–S31. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Holland, P.R.; Martins-Oliveira, M.; Hoffmann, J.; Schankin, C.; Akerman, S. Pathophysiology of Migraine: A Disorder of Sensory Processing. Physiol. Rev. 2017, 97, 553–622. [Google Scholar] [CrossRef]
- Messlinger, K.; Balcziak, L.K.; Russo, A.F. Cross-talk signaling in the trigeminal ganglion: Role of neuropeptides and other mediators. J. Neural Transm. (Vienna) 2020, 127, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Tajti, J.; Uddman, R.; Moller, S.; Sundler, F.; Edvinsson, L. Messenger molecules and receptor mRNA in the human trigeminal ganglion. J. Auton. Nerv. Syst. 1999, 76, 176–183. [Google Scholar] [CrossRef]
- Brouns, I.; Pintelon, I.; Van Genechten, J.; De Proost, I.; Timmermans, J.P.; Adriaensen, D. Vesicular glutamate transporter 2 is expressed in different nerve fibre populations that selectively contact pulmonary neuroepithelial bodies. Histochem. Cell Biol. 2004, 121, 1–12. [Google Scholar] [CrossRef]
- Keast, J.R.; Stephensen, T.M. Glutamate and aspartate immunoreactivity in dorsal root ganglion cells supplying visceral and somatic targets and evidence for peripheral axonal transport. J. Comp. Neurol. 2000, 424, 577–587. [Google Scholar] [CrossRef]
- Dodick, D.; Silberstein, S. Central sensitization theory of migraine: Clinical implications. Headache 2006, 46 (Suppl. 4), S182–S191. [Google Scholar] [CrossRef]
- Antonaci, F.; Nappi, G.; Galli, F.; Manzoni, G.C.; Calabresi, P.; Costa, A. Migraine and psychiatric comorbidity: A review of clinical findings. J. Headache Pain 2011, 12, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Dresler, T.; Caratozzolo, S.; Guldolf, K.; Huhn, J.I.; Loiacono, C.; Niiberg-Pikksoot, T.; Puma, M.; Sforza, G.; Tobia, A.; Ornello, R.; et al. Understanding the nature of psychiatric comorbidity in migraine: A systematic review focused on interactions and treatment implications. J. Headache Pain 2019, 20, 51. [Google Scholar] [CrossRef] [Green Version]
- Juang, K.D.; Wang, S.J.; Fuh, J.L.; Lu, S.R.; Su, T.P. Comorbidity of depressive and anxiety disorders in chronic daily headache and its subtypes. Headache 2000, 40, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Moriwaki, K.; Oiso, H.; Ishigooka, J. High prevalence of comorbidity of migraine in outpatients with panic disorder and effectiveness of psychopharmacotherapy for both disorders: A retrospective open label study. Psychiatry Res. 2011, 185, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Chen, P.K.; Fuh, J.L. Comorbidities of migraine. Front. Neurol. 2010, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- Swanson, S.A.; Zeng, Y.; Weeks, M.; Colman, I. The contribution of stress to the comorbidity of migraine and major depression: Results from a prospective cohort study. BMJ Open 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldacci, F.; Lucchesi, C.; Cafalli, M.; Poletti, M.; Ulivi, M.; Vedovello, M.; Giuntini, M.; Mazzucchi, S.; Del Prete, E.; Vergallo, A.; et al. Migraine features in migraineurs with and without anxiety-depression symptoms: A hospital-based study. Clin. Neurol. Neurosurg. 2015, 132, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Smitherman, T.A.; Maizels, M.; Penzien, D.B. Headache chronification: Screening and behavioral management of comorbid depressive and anxiety disorders. Headache 2008, 48, 45–50. [Google Scholar] [CrossRef]
- Ligthart, L.; Gerrits, M.M.; Boomsma, D.I.; Penninx, B.W. Anxiety and depression are associated with migraine and pain in general: An investigation of the interrelationships. J. Pain 2013, 14, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Grassini, S.; Nordin, S. Comorbidity in Migraine with Functional Somatic Syndromes, Psychiatric Disorders and Inflammatory Diseases: A Matter of Central Sensitization? Behav. Med. 2017, 43, 91–99. [Google Scholar] [CrossRef]
- Pellegrino, A.B.W.; Davis-Martin, R.E.; Houle, T.T.; Turner, D.P.; Smitherman, T.A. Perceived triggers of primary headache disorders: A meta-analysis. Cephalalgia 2018, 38, 1188–1198. [Google Scholar] [CrossRef]
- Peres, M.F.; Sanchez del Rio, M.; Seabra, M.L.; Tufik, S.; Abucham, J.; Cipolla-Neto, J.; Silberstein, S.D.; Zukerman, E. Hypothalamic involvement in chronic migraine. J. Neurol. Neurosurg. Psychiatry 2001, 71, 747–751. [Google Scholar] [CrossRef] [Green Version]
- Woldeamanuel, Y.W.; Sanjanwala, B.M.; Cowan, R.P. Endogenous glucocorticoids may serve as biomarkers for migraine chronification. Ther. Adv. Chronic Dis. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Sauro, K.M.; Becker, W.J. The stress and migraine interaction. Headache 2009, 49, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Hayasaki, H.; Sohma, Y.; Kanbara, K.; Maemura, K.; Kubota, T.; Watanabe, M. A local GABAergic system within rat trigeminal ganglion cells. Eur. J. Neurosci. 2006, 23, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Storer, R.J.; Akerman, S.; Goadsby, P.J. GABA receptors modulate trigeminovascular nociceptive neurotransmission in the trigeminocervical complex. Br. J. Pharmacol. 2001, 134, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Eriksen, H.R.; Ursin, H. Subjective health complaints, sensitization, and sustained cognitive activation (stress). J. Psychosom. Res. 2004, 56, 445–448. [Google Scholar] [CrossRef]
- Morimoto, K.; Fahnestock, M.; Racine, R.J. Kindling and status epilepticus models of epilepsy: Rewiring the brain. Prog. Neurobiol. 2004, 73, 1–60. [Google Scholar] [CrossRef]
- Vieira, D.S.; Naffah-Mazacoratti, M.G.; Zukerman, E.; Senne Soares, C.A.; Alonso, E.O.; Faulhaber, M.H.; Cavalheiro, E.A.; Peres, M.F. Cerebrospinal fluid GABA levels in chronic migraine with and without depression. Brain Res. 2006, 1090, 197–201. [Google Scholar] [CrossRef]
- Henderson, L.A.; Peck, C.C.; Petersen, E.T.; Rae, C.D.; Youssef, A.M.; Reeves, J.M.; Wilcox, S.L.; Akhter, R.; Murray, G.M.; Gustin, S.M. Chronic pain: Lost inhibition? J. Neurosci. 2013, 33, 7574–7582. [Google Scholar] [CrossRef] [Green Version]
- Peek, A.L.; Rebbeck, T.; Puts, N.A.; Watson, J.; Aguila, M.R.; Leaver, A.M. Brain GABA and glutamate levels across pain conditions: A systematic literature review and meta-analysis of 1H-MRS studies using the MRS-Q quality assessment tool. Neuroimage 2020, 210, 116532. [Google Scholar] [CrossRef]
- Spitzer, N.C. How GABA generates depolarization. J. Physiol. 2010, 588, 757–758. [Google Scholar] [CrossRef]
- Gao, Y.; Zhou, J.J.; Zhu, Y.; Kosten, T.; Li, D.P. Chronic Unpredictable Mild Stress Induces Loss of GABA Inhibition in Corticotrophin-Releasing Hormone-Expressing Neurons through NKCC1 Upregulation. Neuroendocrinology 2017, 104, 194–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, S.A.; Wamsteeker, J.I.; Kurz, E.U.; Bains, J.S. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat. Neurosci. 2009, 12, 438–443. [Google Scholar] [CrossRef]
- Sarkar, J.; Wakefield, S.; MacKenzie, G.; Moss, S.J.; Maguire, J. Neurosteroidogenesis is required for the physiological response to stress: Role of neurosteroid-sensitive GABAA receptors. J. Neurosci. 2011, 31, 18198–18210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, B.; Kumada, T.; Furukawa, T.; Inoue, K.; Watanabe, M.; Sato, K.; Fukuda, A. Pre- and post-synaptic switches of GABA actions associated with Cl- homeostatic changes are induced in the spinal nucleus of the trigeminal nerve in a rat model of trigeminal neuropathic pain. Neuroscience 2013, 228, 334–348. [Google Scholar] [CrossRef]
- Jenck, F.; Moreau, J.L.; Martin, J.R. Dorsal periaqueductal gray-induced aversion as a simulation of panic anxiety: Elements of face and predictive validity. Psychiatry Res. 1995, 57, 181–191. [Google Scholar] [CrossRef]
- Seligman, M.E. Learned helplessness. Annu. Rev. Med. 1972, 23, 407–412. [Google Scholar] [CrossRef]
- Hammack, S.E.; Cooper, M.A.; Lezak, K.R. Overlapping neurobiology of learned helplessness and conditioned defeat: Implications for PTSD and mood disorders. Neuropharmacology 2012, 62, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, L.; Edwards, E.; Henn, F.A. Dexamethasone suppression test in helpless rats. Biol. Psychiatry 1989, 26, 530–532. [Google Scholar] [CrossRef]
- Menke, A. Is the HPA Axis as Target for Depression Outdated, or Is There a New Hope? Front. Psychiatry 2019, 10, 101. [Google Scholar] [CrossRef]
- Holsboer, F.; Ising, M. Central CRH system in depression and anxiety--evidence from clinical studies with CRH1 receptor antagonists. Eur. J. Pharmacol. 2008, 583, 350–357. [Google Scholar] [CrossRef]
- Clark, A.J.; Forfar, R.; Hussain, M.; Jerman, J.; McIver, E.; Taylor, D.; Chan, L. ACTH Antagonists. Front. Endocrinol. (Lausanne) 2016, 7, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.J.L.; Chan, L. Stability and Turnover of the ACTH Receptor Complex. Front. Endocrinol. (Lausanne) 2019, 10, 491. [Google Scholar] [CrossRef] [PubMed]
- Durham, P.L.; Cady, R. Insights into the mechanism of onabotulinumtoxinA in chronic migraine. Headache 2011, 51, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, G.P.; Grosberg, B.M.; McAllister, P.J.; Lipton, R.B.; Buse, D.C. Prophylactic onabotulinumtoxinA in patients with chronic migraine and comorbid depression: An open-label, multicenter, pilot study of efficacy, safety and effect on headache-related disability, depression, and anxiety. Int. J. Gen. Med. 2015, 8, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, H.; Wei, Y.; Lian, Y.; Chen, Y.; Zheng, Y. Treatment of chronic daily headache with comorbid anxiety and depression using botulinum toxin A: A prospective pilot study. Int. J. Neurosci. 2017, 127, 285–290. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Damier, P.; Lemonnier, E. Failure of the Nemo Trial: Bumetanide Is a Promising Agent to Treat Many Brain Disorders but Not Newborn Seizures. Front. Cell. Neurosci. 2016, 10, 90. [Google Scholar] [CrossRef] [Green Version]
- Eftekhari, S.; Salvatore, C.A.; Johansson, S.; Chen, T.B.; Zeng, Z.; Edvinsson, L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res. 2015, 1600, 93–109. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalkman, H.O. Stress-Induced Alteration in Chloride Transporters in the Trigeminal Nerve May Explain the Comorbidity between Depression and Migraine. Psychiatry Int. 2020, 1, 36-41. https://doi.org/10.3390/psychiatryint1020006
Kalkman HO. Stress-Induced Alteration in Chloride Transporters in the Trigeminal Nerve May Explain the Comorbidity between Depression and Migraine. Psychiatry International. 2020; 1(2):36-41. https://doi.org/10.3390/psychiatryint1020006
Chicago/Turabian StyleKalkman, Hans O. 2020. "Stress-Induced Alteration in Chloride Transporters in the Trigeminal Nerve May Explain the Comorbidity between Depression and Migraine" Psychiatry International 1, no. 2: 36-41. https://doi.org/10.3390/psychiatryint1020006
APA StyleKalkman, H. O. (2020). Stress-Induced Alteration in Chloride Transporters in the Trigeminal Nerve May Explain the Comorbidity between Depression and Migraine. Psychiatry International, 1(2), 36-41. https://doi.org/10.3390/psychiatryint1020006