
Hypoxia, Acidification and Inflammation: Partners in Crime in Parkinson’s Disease Pathogenesis?


{kind=link}
Abstract
:1. Parkinson’s Disease—A Very Brief Background
2. Regulation of Oxygen Levels and Acidity in the Brain
2.1. Oxygen-Sensing and Consumption in PD Brain
2.2. pH in PD Brain
3. Inflammation and pH Alterations in Brain Aging
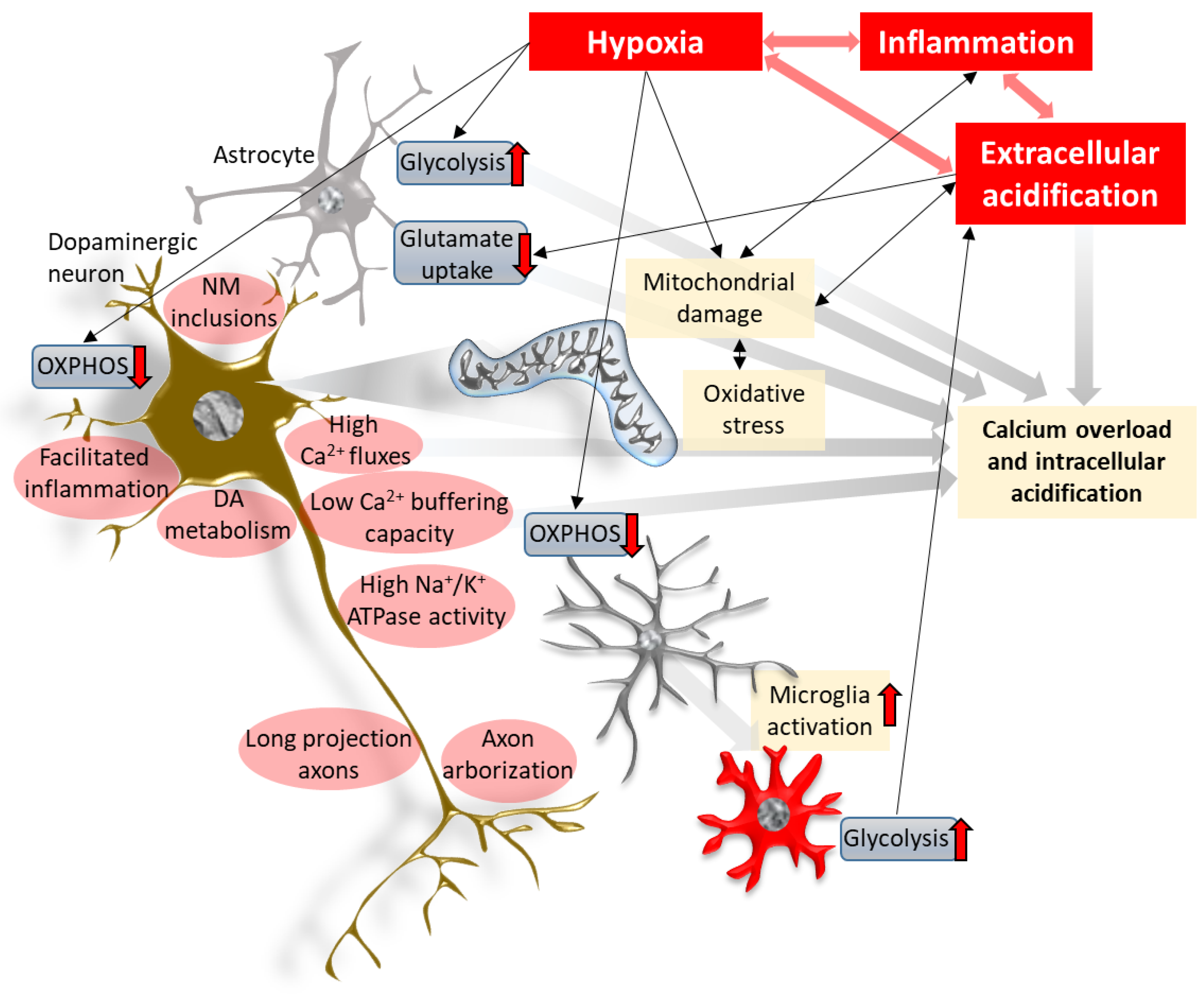
4. The Interplay between Hypoxia, Acidification and Inflammation
Author Contributions
Funding
Conflicts of Interest
References
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S. Description of Parkinson’s disease as a clinical syndrome. Ann. N. Y. Acad. Sci. 2003, 991, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The role of mitochondria in neurodegenerative diseases: The lesson from Alzheimer’s disease and Parkinson’s disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions About Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2012, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated Energy Budgets for Neural Computation in the Neocortex and Cerebellum. J. Cereb. Blood Flow Metab. 2012, 32, 1222–1232. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, F.A.C.; Carvalho, L.R.B.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.L.; Leite, R.E.P.; Filho, W.J.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Oorschot, D.E. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: A stereological study using the cavalieri and optical disector methods. J. Comp. Neurol. 1996, 366, 580–599. [Google Scholar] [CrossRef]
- Roberts, R.C.; Force, M.; Kung, L. Dopaminergic synapses in the matrix of the ventrolateral striatum after chronic haloperidol treatment. Synapse 2002, 45, 78–85. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single Nigrostriatal Dopaminergic Neurons Form Widely Spread and Highly Dense Axonal Arborizations in the Neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Parent, M.; Parent, A. Relationship between axonal collateralization and neuronal degeneration in basal ganglia. In Parkinson’s Disease and Related Disorders; Springer: Vienna, Austria, 2006; pp. 85–88. [Google Scholar]
- Braak, H.; Bohl, J.R.; Müller, C.M.; Rüb, U.; de Vos, R.A.; Del Tredici, K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord. 2006, 21, 2042–2051. [Google Scholar] [CrossRef]
- Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007, 30, 244–250. [Google Scholar] [CrossRef]
- Seutin, V.; Shen, K.-Z.; North, R.; Johnson, S. Sulfonylurea-sensitive potassium current evoked by sodium-loading in rat midbrain dopamine neurons. Neuroscience 1996, 71, 709–719. [Google Scholar] [CrossRef]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Tan, E.-K.; Chao, Y.-X.; West, A.; Chan, L.-L.; Poewe, W.; Jankovic, J. Parkinson disease and the immune system—Associations, mechanisms and therapeutics. Nat. Rev. Neurol. 2020, 16, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 1978, 14, 633–643. [Google Scholar]
- Sulzer, D.; Zecca, L. Intraneuronal dopamine-quinone synthesis: A review. Neurotox. Res. 1999, 1, 181–195. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V.R. ROS and Parkinson’s Disease: A View to Kill. In Free Radicals in Brain Pathophysiology; Taylor & Francis Group: Boca Raton, FL, USA, 2003. [Google Scholar]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Syed, M.M.K.; Lashuel, H.A.; Millet, G.P. Hypoxia Conditioning as a Promising Therapeutic Target in Parkinson’s Disease? Mov. Disord. 2021, 36, 857–861. [Google Scholar] [CrossRef]
- Pokusa, M.; Hajdúchová, D.; Buday, T.; Kralova Trancikova, A. Respiratory Function and Dysfunction in Parkinson-Type Neurodegeneration. Physiol. Res. 2020, 69, S69–S79. [Google Scholar] [CrossRef]
- Vijayan, S.; Singh, B.; Ghosh, S.; Stell, R.; Mastaglia, F.L. Brainstem ventilatory dysfunction: A plausible mechanism for dyspnea in Parkinson’s Disease? Mov. Disord. 2020, 35, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Onodera, H.; Okabe, S.; Kikuchi, Y.; Tsuda, T.; Itoyama, Y. Impaired chemosensitivity and perception of dyspnoea in Parkinson’s disease. Lancet 2000, 356, 739–740. [Google Scholar] [CrossRef]
- Benarroch, E.E.; Schmeichel, A.M.; Low, P.A.; Parisi, J.E. Depletion of putative chemosensitive respiratory neurons in the ventral medullary surface in multiple system atrophy. Brain 2007, 130, 469–475. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of Mammalian O2 Homeostasis by Hypoxia-Inducible Factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef] [PubMed]
- Heras-Garvin, A.; Msc, C.D.; Eschlböck, S.; Holton, J.L.; Wenning, G.K.; Stefanova, N. Signs of Chronic Hypoxia Suggest a Novel Pathophysiological Event in α-Synucleinopathies. Mov. Disord. 2020, 35, 2333–2338. [Google Scholar] [CrossRef]
- Zakharova, E.T.; Sokolov, A.V.; Pavlichenko, N.N.; Kostevich, V.A.; Abdurasulova, I.N.; Chechushkov, A.V.; Voynova, I.V.; Elizarova, A.Y.; Kolmakov, N.N.; Bass, M.G.; et al. Erythropoietin and Nrf2: Key factors in the neuroprotection provided by apo-lactoferrin. BioMetals 2018, 31, 425–443. [Google Scholar] [CrossRef]
- Xu, S.-F.; Zhang, Y.-H.; Wang, S.; Pang, Z.-Q.; Fan, Y.-G.; Li, J.-Y.; Wang, Z.-Y.; Guo, C. Lactoferrin ameliorates dopaminergic neurodegeneration and motor deficits in MPTP-treated mice. Redox Biol. 2019, 21, 101090. [Google Scholar] [CrossRef]
- Guo, C.; Hao, L.-J.; Yang, Z.-H.; Chai, R.; Zhang, S.; Gu, Y.; Gao, H.-L.; Zhong, M.-L.; Wang, T.; Li, J.-Y.; et al. Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp. Neurol. 2016, 280, 13–23. [Google Scholar] [CrossRef]
- Qin, L.; Shu, L.; Zhong, J.; Pan, H.; Guo, J.; Sun, Q.; Yan, X.; Tang, B.; Xu, Q. Association of HIF1A and Parkinson’s disease in a Han Chinese population demonstrated by molecular inversion probe analysis. Neurol. Sci. 2019, 40, 1927–1931. [Google Scholar] [CrossRef]
- Rehncrona, S. Brain acidosis. Ann. Emerg. Med. 1985, 14, 770–776. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Goncalves, R.L.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deitmer, J. Acid-base transport and pH regulation. Handb. Neurochem. Mol. Neurobiol. 2007, 5, 469–486. [Google Scholar]
- Swietach, P.; Hulikova, A.; Vaughan-Jones, R.D.; Harris, A.L. New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene 2010, 29, 6509–6521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagadicgossmann, D.; Huc, L.; Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: Origins and roles. Cell Death Differ. 2004, 11, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, C.L.; Leong, S.L.; Ali, F.E.; Kenche, V.B.; Hill, A.F.; Gras, S.L.; Barnham, K.J.; Cappai, R. Dopamine and the dopamine oxidation product 5, 6-dihydroxylindole promote distinct on-pathway and off-pathway aggregation of α-synuclein in a pH-dependent manner. J. Mol. Biol. 2009, 387, 771–785. [Google Scholar] [CrossRef]
- Umek, N.; Geršak, B.; Vintar, N.; Šoštarič, M.; Mavri, J. Dopamine Autoxidation Is Controlled by Acidic pH. Front. Mol. Neurosci. 2018, 11, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Moreno, M.; Rodríguez-López, J.; Martínez-Ortiz, F.; Tudela, J.; Varón, R.; García-Cánovas, F. Effect of pH on the oxidation pathway of dopamine catalyzed by tyrosinase. Arch. Biochem. Biophys. 1991, 288, 427–434. [Google Scholar] [CrossRef]
- Wemmie, J.A.; Price, M.P.; Welsh, M.J. Acid-sensing ion channels: Advances, questions and therapeutic opportunities. Trends Neurosci. 2006, 29, 578–586. [Google Scholar] [CrossRef]
- Wemmie, J.A.; Taugher, R.J.; Kreple, C.J. Acid-sensing ion channels in pain and disease. Nat. Rev. Neurosci. 2013, 14, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatck, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Kuznetsov, A.S.; Kopell, N.J.; Wilson, C.J. Transient High-Frequency Firing in a Coupled-Oscillator Model of the Mesencephalic Dopaminergic Neuron. J. Neurophysiol. 2006, 95, 932–947. [Google Scholar] [CrossRef] [PubMed]
- Foehring, R.C.; Zhang, X.F.; Lee, J.; Callaway, J.C. Endogenous Calcium Buffering Capacity of Substantia Nigral Dopamine Neurons. J. Neurophysiol. 2009, 102, 2326–2333. [Google Scholar] [CrossRef] [Green Version]
- Nagley, P.; Higgins, G.C.; Atkin, J.D.; Beart, P.M. Multifaceted deaths orchestrated by mitochondria in neurones. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 167–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, R.L.; Sung, M.-L.A.; Vasylyev, D.; Zhang, M.-Y.; Albinson, K.; Kubek, K.; Kagan, N.; Beyer, C.; Lin, Q.; Dwyer, J.M.; et al. Amiloride is neuroprotective in an MPTP model of Parkinson’s disease. Neurobiol. Dis. 2008, 31, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Komnig, D.; Imgrund, S.; Reich, A.; Gründer, S.; Falkenburger, B.H. ASIC1a Deficient Mice Show Unaltered Neurodegeneration in the Subacute MPTP Model of Parkinson Disease. PLoS ONE 2016, 11, e0165235. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Martinez-Vicente, M.; Caldwell, G.A.; Caldwell, K.A.; Yue, Z.; Cookson, M.R.; Klein, C.; Vila, M.; Bezard, E. Lysosomal impairment in Parkinson’s disease. Mov. Disord. 2013, 28, 725–732. [Google Scholar] [CrossRef] [PubMed]
- McClendon, S.; Rospigliosi, C.C.; Eliezer, D. Charge neutralization and collapse of the C-terminal tail of alpha-synuclein at low pH. Protein Sci. 2009, 18, 1531–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Cole, N.B.; DiEuliis, D.; Leo, P.; Mitchell, D.C.; Nussbaum, R.L. Mitochondrial translocation of α-synuclein is promoted by intracellular acidification. Exp. Cell Res. 2008, 314, 2076–2089. [Google Scholar] [CrossRef] [Green Version]
- Bowen, B.C.; Block, R.E.; Sanchez-Ramos, J.; Pattany, P.M.; Lampman, D.A.; Murdoch, J.B.; Quencer, R.M. Proton MR spectroscopy of the brain in 14 patients with Parkinson disease. Am. J. Neuroradiol. 1995, 16, 61–68. [Google Scholar]
- Firbank, M.J.; Harrison, R.M.; O’Brien, J.T. A comprehensive review of proton magnetic resonance spectroscopy studies in dementia and Parkinson’s disease. Dement. Geriatr. Cogn. Disord. 2002, 14, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.-W.; Liao, K.-F.; Lin, C.-L.; Lin, C.-H. Association between Parkinson’s disease and proton pump inhibitors therapy in older people. Biomedicine 2020, 10, 1–4. [Google Scholar] [PubMed]
- Nielsen, H.H.; Qiu, J.; Friis, S.; Wermuth, L.; Ritz, B. Treatment for Helicobacter pylori infection and risk of parkinson’s disease in Denmark. Eur. J. Neurol. 2012, 19, 864–869. [Google Scholar] [CrossRef] [Green Version]
- Pirchl, M.; Marksteiner, J.; Humpel, C. Effects of acidosis on brain capillary endothelial cells and cholinergic neurons: Relevance to vascular dementia and Alzheimer’s disease. Neurol. Res. 2006, 28, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Wang, D.; Huang, M.; Yu, G.; Li, H. Hypothesis on the relationship between the change in intracellular pH and incidence of sporadic Alzheimer’s disease or vascular dementia. Int. J. Neurosci. 2010, 120, 591–595. [Google Scholar] [CrossRef]
- Kuo, S.-W.; Jiang, M.; Heckman, C. Potential involvement of intracellular pH in a mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 15, 151–153. [Google Scholar] [CrossRef] [Green Version]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s disease: Different sides of the same coin? Mov. Disord. 2017, 32, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Polonis, K.; Becari, C.; Chahal, C.A.A.; Zhang, Y.; Allen, A.M.; Kellogg, T.A.; Somers, V.K.; Singh, P. Chronic Intermittent Hypoxia Triggers a Senescence-like Phenotype in Human White Preadipocytes. Sci. Rep. 2020, 10, 6846. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Hypoxia-Inducible Histone Lysine Demethylases: Impact on the Aging Process and Age-Related Diseases. Aging Dis. 2016, 7, 180–200. [Google Scholar]
- Wilson, E.N.; Anderson, M.; Snyder, B.; Duong, P.; Trieu, J.; Schreihofer, D.A.; Cunningham, R.L. Chronic intermittent hypoxia induces hormonal and male sexual behavioral changes: Hypoxia as an advancer of aging. Physiol. Behav. 2018, 189, 64–73. [Google Scholar] [CrossRef]
- Kim, H.; Lee, D.-K.; Choi, J.-W.; Kim, J.-S.; Park, S.C.; Youn, H.-D. Analysis of the effect of aging on the response to hypoxia by cDNA microarray. Mech. Ageing Dev. 2003, 124, 941–949. [Google Scholar] [CrossRef]
- Frenkel-Denkberg, G.; Gershon, D.; Levy, A.P. The function of hypoxia-inducible factor 1 (HIF-1) is impaired in senescent mice. FEBS Lett. 1999, 462, 341–344. [Google Scholar] [CrossRef] [Green Version]
- Kayser, E.-B.; Sedensky, M.M.; Morgan, P.G.; Hoppel, C.L. Mitochondrial Oxidative Phosphorylation Is Defective in the Long-lived Mutant clk-1. J. Biol. Chem. 2004, 279, 54479–54486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Bussière, F.; Hekimi, S. Mitochondrial Electron Transport Is a Key Determinant of Life Span in Caenorhabditis elegans. Dev. Cell 2001, 1, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Lee, R.Y.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 2003, 33, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Copeland, J.M.; Cho, J.; Lo, T., Jr.; Hur, J.H.; Bahadorani, S.; Arabyan, T.; Rabie, J.; Soh, J.; Walker, D.W. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 2009, 19, 1591–1598. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, J.; Hekimi, S. Early mitochondrial dysfunction in long-lived Mclk1+/-mice. J. Biol. Chem. 2008, 283, 26217–26227. [Google Scholar] [CrossRef] [Green Version]
- Dell’Agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Burtscher, J.; Mallet, R.T.; Burtscher, M.; Millet, G.P. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing Res. Rev. 2021, 68, 101343. [Google Scholar] [CrossRef] [PubMed]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.-W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Minakaki, G.; Krainc, D.; Burbulla, L.F. The Convergence of Alpha-Synuclein, Mitochondrial, and Lysosomal Pathways in Vulnerability of Midbrain Dopaminergic Neurons in Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 580634. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, S.S.; Petersen, D.; Marlet, F.R.; Kücükköse, E.; Galvagnion, C. The interplay between GCase, α-synuclein and lipids in human models of Parkinson’s disease. Biophys. Chem. 2020, 273, 106534. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Verona, G.; Schapira, A.H. Glucocerebrosidase deficiency promotes release of α-synuclein fibrils from cultured neurons. Hum. Mol. Genet. 2020, 29, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Torres, C. Faculty Opinions recommendation of Astrocyte senescence: Evidence and significance. Fac. Opin. Post Publ. Peer Rev. Biomed. Lit. 2019, 18, e12937. [Google Scholar]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.-Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; DeMaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Majdi, A.; Mahmoudi, J.; Sadigh-Eteghad, S.; Golzari, S.E.; Sabermarouf, B.; Reyhani-Rad, S. Permissive role of cytosolic pH acidification in neurodegeneration: A closer look at its causes and consequences. J. Neurosci. Res. 2016, 94, 879–887. [Google Scholar] [CrossRef]
- Swenson, E.R. Hypoxia and Its Acid–Base Consequences: From Mountains to Malignancy. In Hypoxia: Translation in Progress; Roach, R.C., Hackett, P.H., Wagner, P.D., Eds.; Springer: Boston, MA, USA, 2016; pp. 301–323. [Google Scholar]
- Ivashkiv, L.B. The hypoxia–lactate axis tempers inflammation. Nat. Rev. Immunol. 2020, 20, 85–86. [Google Scholar] [CrossRef]
- Davies, A.L.; Desai, R.A.; Bloomfield, P.S.; McIntosh, P.R.; Chapple, K.J.; Linington, C.; Fairless, R.; Diem, R.; Kasti, M.; Murphy, M.P.; et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann. Neurol. 2013, 74, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Okajima, F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell. Signal. 2013, 25, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nat. Cell Biol. 2015, 520, 553–557. [Google Scholar]
- Rajamäki, K.; Nordström, T.; Nurmi, K.; Åkerman, K.E.O.; Kovanen, P.T.; Öörni, K.; Eklund, K.K. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J. Biol. Chem. 2013, 288, 13410–13419. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.; Basit, F.; Swarts, H.G.; Forkink, M.; Oliveira, P.J.; Willems, P.H.G.M.; Koopman, W.J.H. Extracellular acidification induces ROS- and mPTP-mediated death in HEK293 cells. Redox Biol. 2018, 15, 394–404. [Google Scholar] [CrossRef]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Wang, M.-X.; Cheng, X.-Y.; Jin, M.; Cao, Y.-L.; Yang, Y.-P.; Wang, J.-D.; Li, Q.; Wang, F.; Hu, L.-F.; Liu, C.-F. TNF compromises lysosome acidification and reduces α-synuclein degradation via autophagy in dopaminergic cells. Exp. Neurol. 2015, 271, 112–121. [Google Scholar] [CrossRef]
- Bu, X.-L.; Wang, X.; Xiang, Y.; Zeng, F.; Liu, Y.-H.; Xiang, Y.; Liang, C.-R.; Wang, Q.H.; Wang, X.; Cao, H.-Y.; et al. The association between infectious burden and Parkinson’s disease: A case-control study. Parkinsonism Relat. Disord. 2015, 21, 877–881. [Google Scholar] [CrossRef]
- Piancone, F.; Saresella, M.; La Rosa, F.; Marventano, I.; Meloni, M.; Navarro, J.; Clerici, M. Inflammatory Responses to Monomeric and Aggregated α-Synuclein in Peripheral Blood of Parkinson Disease Patients. Front. Neurosci. 2021, 15, 235. [Google Scholar] [CrossRef]
- Harms, A.S.; Delic, V.; Thome, A.D.; Bryant, N.; Liu, Z.; Chandra, S.; Jurkuvenaite, A.; West, A.B. α-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol. Commun. 2017, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Tieu, K.; Choi, D.-K.; Wu, D.-C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindå, H.; Hammarberg, H.; Piehl, F.; Khademi, M.; Olsson, T. Expression of MHC class I heavy chain and β2-microglobulin in rat brainstem motoneurons and nigral dopaminergic neurons. J. Neuroimmunol. 1999, 101, 76–86. [Google Scholar] [CrossRef]
- Xu, J.; Peng, Z.; Li, R.; Dou, T.; Xu, W.; Gu, G.; Liu, Y.; Kang, Z.; Tao, H.; Zhang, J.H.; et al. Normoxic induction of cerebral HIF-1α by acetazolamide in rats: Role of acidosis. Neurosci. Lett. 2009, 451, 274–278. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; O’Neill, L.A. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF Transcription Factors, Inflammation, and Immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Watts, E.R.; Walmsley, S.R. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol. Med. 2019, 25, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, G.; Xu, Z.-G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189. [Google Scholar] [CrossRef]
- Bender, A.S.; Young, L.P.; Norenberg, M.D. Effect of lactic acid on l-glutamate uptake in cultured astrocytes: Mechanistic considerations. Brain Res. 1997, 750, 59–66. [Google Scholar] [CrossRef]
- Xiang, Z.; Yuan, M.; Hassen, G.W.; Gampel, M.; Bergold, P.J. Lactate induced excitotoxicity in hippocampal slice cultures. Exp. Neurol. 2004, 186, 70–77. [Google Scholar] [CrossRef]
- Mills, E.; O’Neill, L.A. Succinate: A metabolic signal in inflammation. Trends Mol. Med. 2014, 24, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, S. A Novel, Multi-Faceted Perception of Lactate in Neurology. Front. Neurosci. 2020, 14, 460. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Lei, H.; Thevenet, J.; Gruetter, R.; Magistretti, P.J.; Hirt, L. Neuroprotective Role of Lactate after Cerebral Ischemia. Br. J. Pharmacol. 2009, 29, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Harguindey, S.; Stanciu, D.; Devesa, J.; Alfarouk, K.; Cardone, R.A.; Orozco, J.D.P.; Devesa, P.; Rauch, C.; Orive, G.; Anitua, E.; et al. Cellular acidification as a new approach to cancer treatment and to the understanding and therapeutics of neurodegenerative diseases. Semin. Cancer Biol. 2017, 43, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Chaib, M.; Rathmell, J.C. Immunometabolism: From basic mechanisms to translation. Immunol. Rev. 2020, 295, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lercher, A.; Baazim, H.; Bergthaler, A. Systemic Immunometabolism: Challenges and Opportunities. Immunity 2020, 53, 496–509. [Google Scholar] [CrossRef]
- Wang, A.; Luan, H.H.; Medzhitov, R. An evolutionary perspective on immunometabolism. Science 2019, 363, eaar3932. [Google Scholar] [CrossRef]
- Mallet, R.T.; Burtscher, J.; Manukhina, E.B.; Downey, H.F.; Glazachev, O.S.; Serebrovskaya, T.V.; Burtscher, M. Hypoxic–hyperoxic conditioning and dementia. In Diagnosis and Management in Dementia; Elsevier: Amsterdam, The Netherlands, 2020; pp. 745–760. [Google Scholar]
- Burtscher, J.; Maglione, V.; Di Pardo, A.; Millet, G.P.; Schwarzer, C.; Zangrandi, L. A Rationale for Hypoxic and Chemical Conditioning in Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 582. [Google Scholar] [CrossRef]
- Griffith, H.R.; den Hollander, J.A.; Okonkwo, O.C.; O’Brien, T.; Watts, R.L.; Marson, D.C. Brain metabolism differs in Alzheimer’s disease and Parkinson’s disease dementia. Alzheimers Dement. 2008, 4, 421–427. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burtscher, J.; Millet, G.P. Hypoxia, Acidification and Inflammation: Partners in Crime in Parkinson’s Disease Pathogenesis? Immuno 2021, 1, 78-90. https://doi.org/10.3390/immuno1020006
Burtscher J, Millet GP. Hypoxia, Acidification and Inflammation: Partners in Crime in Parkinson’s Disease Pathogenesis? Immuno. 2021; 1(2):78-90. https://doi.org/10.3390/immuno1020006
Chicago/Turabian StyleBurtscher, Johannes, and Grégoire P. Millet. 2021. "Hypoxia, Acidification and Inflammation: Partners in Crime in Parkinson’s Disease Pathogenesis?" Immuno 1, no. 2: 78-90. https://doi.org/10.3390/immuno1020006
APA StyleBurtscher, J., & Millet, G. P. (2021). Hypoxia, Acidification and Inflammation: Partners in Crime in Parkinson’s Disease Pathogenesis? Immuno, 1(2), 78-90. https://doi.org/10.3390/immuno1020006