
A Signaling View into the Inflammatory Tumor Microenvironment



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Tumor Microenvironment (TME)
2.1. Cancer Cells
2.2. Cells of Mesenchymal Origin
2.2.1. Cancer-Associated Fibroblasts (CAFs)
2.2.2. Endothelial Cells (ECs)
2.3. Immune Cells
2.3.1. Tumor-Associated Macrophages (TAMs)
2.3.2. Tumor-Associated Neutrophils (TANs)
2.3.3. T Lymphocytes
2.3.4. B Lymphocytes
2.3.5. Other Immune Cells
2.4. Non-Cellular Components
The Extracellular Matrix (ECM)
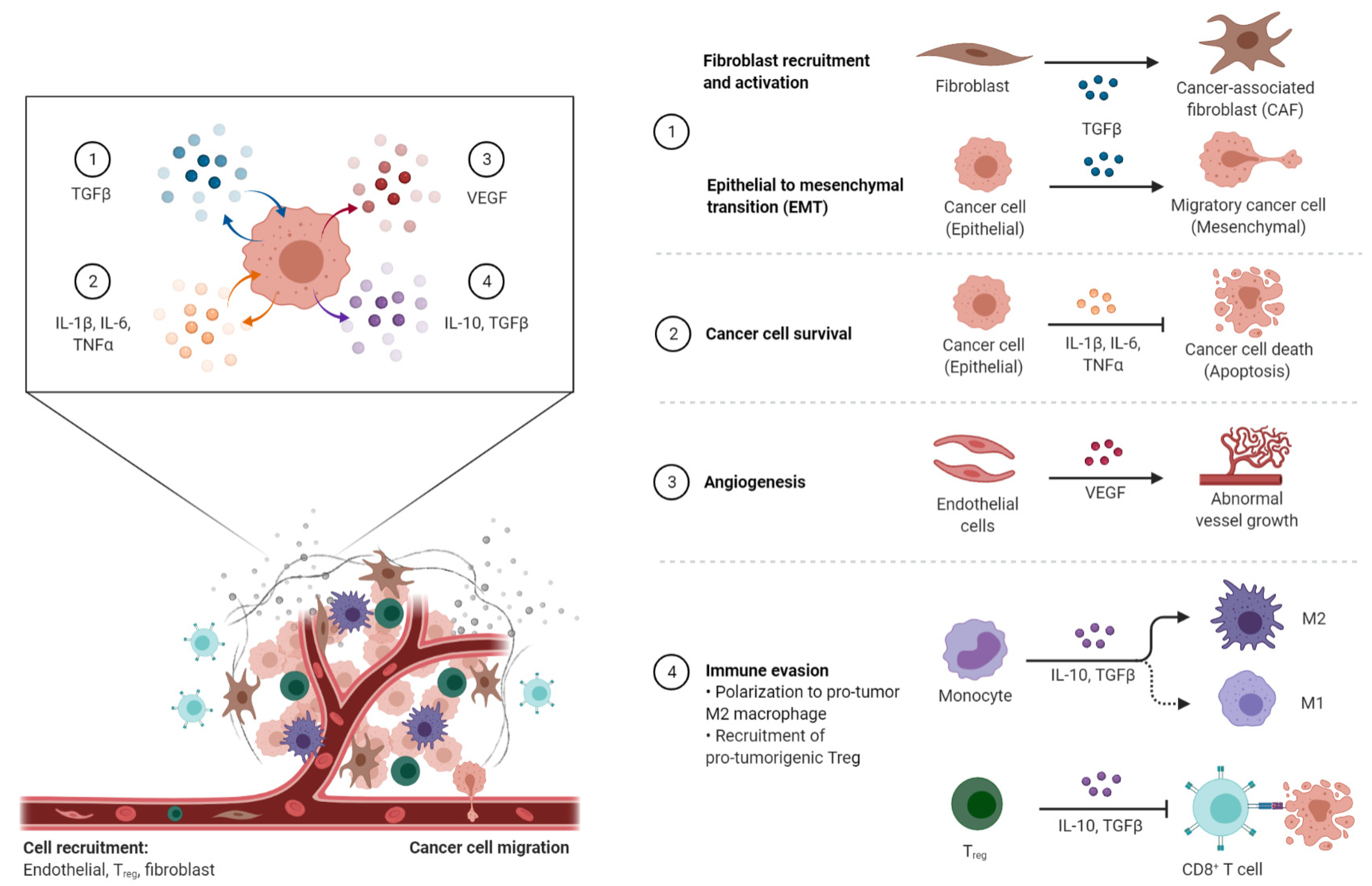
3. Cancer-Associated Inflammation (CAI) and the TME
3.1. Mediators of Cancer-Associated Inflammation (CAI)
3.1.1. Pro-Inflammatory Factors
3.1.2. Pro-Inflammatory Chemokines in the TME
3.1.3. Anti-Inflammatory Mediators—IL-10
3.1.4. The Transforming Growth Factor-β (TGF-β)
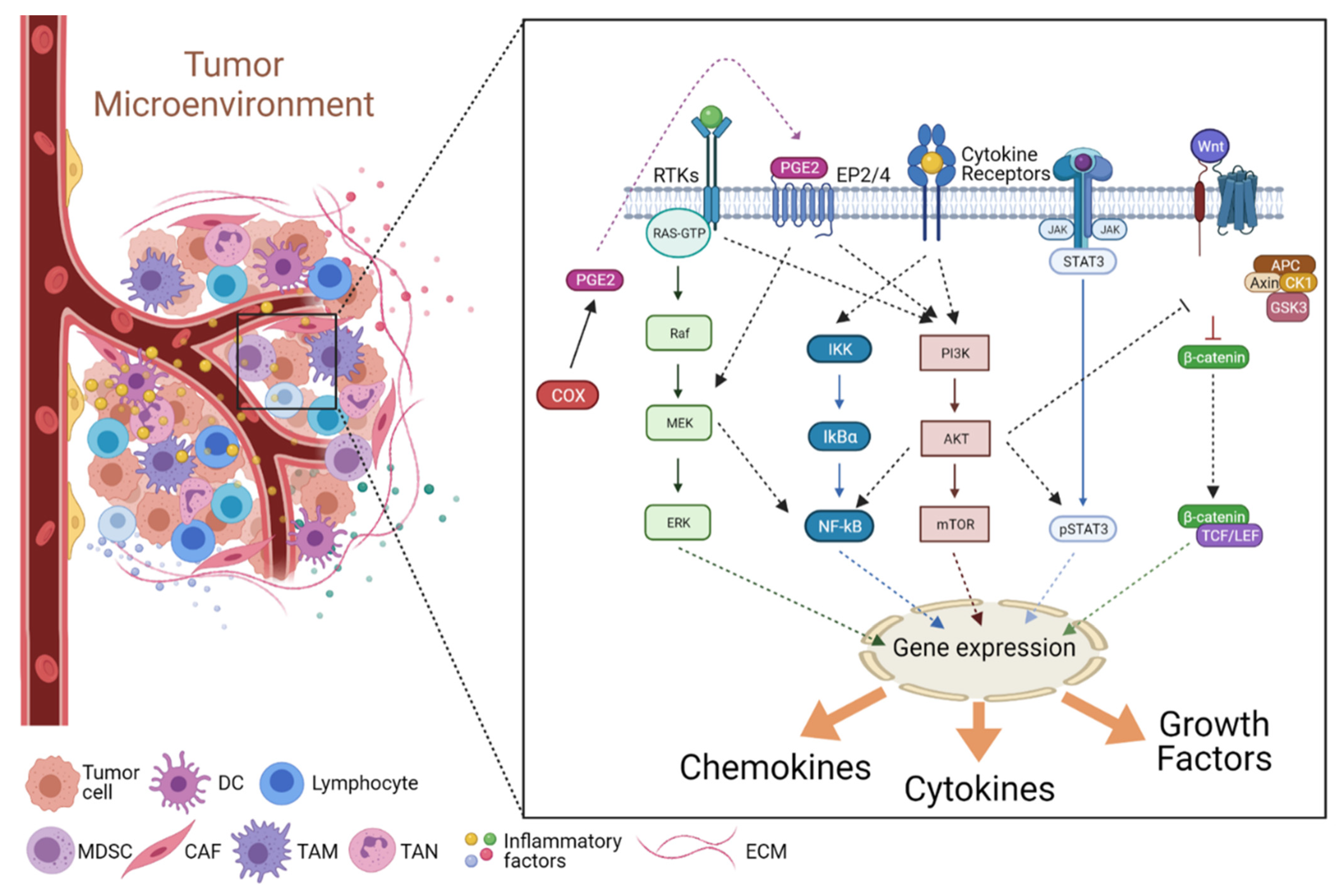
3.2. Signaling in the Inflammatory TME
3.2.1. The JAK/STAT Pathway
3.2.2. The NF-κB Pathway
3.2.3. The COX2/PGE2 Pathway
3.2.4. The PI3K/Akt Pathway
3.2.5. The Wnt Pathway
4. TME in Cancer: Aggressor or Innocent Bystander?
Funding
Conflicts of Interest
References
- Li, L.; Yu, R.; Cai, T.; Chen, Z.; Lan, M.; Zou, T.; Wang, B.; Wang, Q.; Zhao, Y.; Cai, Y. Effects of Immune Cells and Cytokines on Inflammation and Immunosuppression in the Tumor Microenvironment. Int. Immunopharmacol. 2020, 88, 106939. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Fan, X.; Houghton, J. Tumor Microenvironment: The Role of the Tumor Stroma in Cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New Horizons in Tumor Microenvironment Biology: Challenges and Opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of Tumor Microenvironment in Tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the Cancer Stem Cells—What Challenges Do They Pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, F.D.S.E.; Vermeulen, L.; Fessler, E.; Medema, J.P. Cancer Heterogeneity—A Multifaceted View. EMBO Rep. 2013, 14, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almendro, V.; Marusyk, A.; Polyak, K. Cellular Heterogeneity and Molecular Evolution in Cancer. Annu. Rev. Pathol. 2013, 8, 277–302. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. The Cancer Stem Cell: Premises, Promises and Challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Beck, B.; Blanpain, C. Unravelling Cancer Stem Cell Potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Garg, M. Epithelial Plasticity, Autophagy and Metastasis: Potential Modifiers of the Crosstalk to Overcome Therapeutic Resistance. Stem Cell Rev. Rep. 2020, 16, 503–510. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.A.; Laverdet, B.; Bonté, F.; Desmoulière, A. Fibroblasts and Myofibroblasts in Wound Healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in Cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Liu, T.; Yin, R. Biomarkers for Cancer-Associated Fibroblasts. Biomark. Res. 2020, 8, 64. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-Beta Signaling in Fibroblasts Modulates the Oncogenic Potential of Adjacent Epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [Green Version]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The Clinical Role of the TME in Solid Cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Ziani, L.; Safta-Saadoun, T.B.; Gourbeix, J.; Cavalcanti, A.; Robert, C.; Favre, G.; Chouaib, S.; Thiery, J. Melanoma-Associated Fibroblasts Decrease Tumor Cell Susceptibility to NK Cell-Mediated Killing through Matrix-Metalloproteinases Secretion. Oncotarget 2017, 8, 19780–19794. [Google Scholar] [CrossRef] [Green Version]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal Gene Expression Defines Poor-Prognosis Subtypes in Colorectal Cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Franco-Barraza, J.; Francescone, R.; Luong, T.; Shah, N.; Madhani, R.; Cukierman, G.; Dulaimi, E.; Devarajan, K.; Egleston, B.L.; Nicolas, E.; et al. Matrix-Regulated Integrin Avβ5 Maintains A5β1-Dependent Desmoplastic Traits Prognostic of Neoplastic Recurrence. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, R.L.; Puré, E. Cancer-Associated Fibroblasts and Their Influence on Tumor Immunity and Immunotherapy. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döme, B.; Hendrix, M.J.C.; Paku, S.; Tóvári, J.; Tímár, J. Alternative Vascularization Mechanisms in Cancer: Pathology and Therapeutic Implications. Am. J. Pathol. 2007, 170, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Yang, L.; Nie, L.; Lin, H. Unraveling the Molecular Mechanisms between Inflammation and Tumor Angiogenesis. Am. J. Cancer Res. 2021, 11, 301–317. [Google Scholar]
- Jain, R.K. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suva, L.J.; Washam, C.; Nicholas, R.W.; Griffin, R.J. Bone Metastasis: Mechanisms and Therapeutic Opportunities. Nat. Rev. Endocrinol. 2011, 7, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Mohamed, H.T.; El-Husseiny, N.; El Mahdy, M.M.; Safwat, G.; Diab, A.A.; El-Sherif, A.A.; El-Shinawi, M.; Mohamed, M.M. IL-8 Secreted by Tumor Associated Macrophages Contribute to Lapatinib Resistance in HER2-Positive Locally Advanced Breast Cancer via Activation of Src/STAT3/ERK1/2-Mediated EGFR Signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118995. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.-R.; Du, W.-L.; Liu, Y.; Mao, C.-S.; Jin, K.-T.; Yang, X. Role of Immune Regulatory Cells in Breast Cancer: Foe or Friend? Int. Immunopharmacol. 2021, 96, 107627. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Kakolyris, S.; O’Byrne, K.J.; Apostolikas, N.; Skarlatos, J.; Gatter, K.C.; Harris, A.L. Different Patterns of Stromal and Cancer Cell Thymidine Phosphorylase Reactivity in Non-Small-Cell Lung Cancer: Impact on Tumour Neoangiogenesis and Survival. Br. J. Cancer 1998, 77, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dönmez, T.; Höhne, K.; Zissel, G.; Herrmann, K.; Hautzel, H.; Aigner, C.; Hegedüs, B.; Ploenes, T. Insights into Immunometabolism: A Dataset Correlating the 18FDG PET/CT Maximum Standard Uptake Value of the Primary Tumor with the CCL18 Serum Level in Non-Small Cell Lung Cancer. Data Brief 2021, 35, 106859. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.S.; Wright, G.; Tan, B.; Rosenwald, A.; Gascoyne, R.D.; Chan, W.C.; Fisher, R.I.; Braziel, R.M.; Rimsza, L.M.; Grogan, T.M.; et al. Prediction of Survival in Follicular Lymphoma Based on Molecular Features of Tumor-Infiltrating Immune Cells. N. Engl. J. Med. 2004, 351, 2159–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The Role of Tumour-Associated Macrophages in Tumour Progression: Implications for New Anticancer Therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.P.; Zhang, X.; Frauwirth, K.A.; Mosser, D.M. Biochemical and Functional Characterization of Three Activated Macrophage Populations. J. Leukoc. Biol. 2006, 80, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimp, A.H.; de Vries, E.G.E.; Scherphof, G.L.; Daemen, T. A Potential Role of Macrophage Activation in the Treatment of Cancer. Crit. Rev. Oncol. Hematol. 2002, 44, 143–161. [Google Scholar] [CrossRef]
- Swann, J.B.; Vesely, M.D.; Silva, A.; Sharkey, J.; Akira, S.; Schreiber, R.D.; Smyth, M.J. Demonstration of Inflammation-Induced Cancer and Cancer Immunoediting during Primary Tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 652–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, M.E.; Fridlender, Z.G. Tumour-Associated Neutrophils in Patients with Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil Plasticity in the Tumor Microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Zajac, E.; Juncker-Jensen, A.; Kupriyanova, T.A.; Welter, L.; Quigley, J.P. Tissue-Infiltrating Neutrophils Constitute the Major in Vivo Source of Angiogenesis-Inducing MMP-9 in the Tumor Microenvironment. Neoplasia 2014, 16, 771–788. [Google Scholar] [CrossRef] [Green Version]
- Ostanin, D.V.; Kurmaeva, E.; Furr, K.; Bao, R.; Hoffman, J.; Berney, S.; Grisham, M.B. Acquisition of Antigen-Presenting Functions by Neutrophils Isolated from Mice with Chronic Colitis. J. Immunol. 2012, 188, 1491–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B Cell-Helper Neutrophils Stimulate the Diversification and Production of Immunoglobulin in the Marginal Zone of the Spleen. Nat. Immunol. 2011, 13, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Pillay, J.; Kamp, V.M.; van Hoffen, E.; Visser, T.; Tak, T.; Lammers, J.-W.; Ulfman, L.H.; Leenen, L.P.; Pickkers, P.; Koenderman, L. A Subset of Neutrophils in Human Systemic Inflammation Inhibits T Cell Responses through Mac-1. J. Clin. Investig. 2012, 122, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Saini, M.; Szczerba, B.M.; Aceto, N. Circulating Tumor Cell-Neutrophil Tango along the Metastatic Process. Cancer Res. 2019, 79, 6067–6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, S.; Baba, T.; Muranaka, H.; Tanabe, Y.; Takahashi, C.; Matsugo, S.; Mukaida, N. Involvement of Prokineticin 2-Expressing Neutrophil Infiltration in 5-Fluorouracil-Induced Aggravation of Breast Cancer Metastasis to Lung. Mol. Cancer Ther. 2018, 17, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, A.; Garasa, S.; Ochoa, M.C.; Villalba, M.; Olivera, I.; Cirella, A.; Eguren-Santamaria, I.; Berraondo, P.; Schalper, K.A.; de Andrea, C.E.; et al. IL8, Neutrophils, and NETs in a Collusion against Cancer Immunity and Immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Teijeira, Á.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps That Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871.e8. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated Neutrophil Extracellular Traps Limit Inflammation by Degrading Cytokines and Chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Clemente, C.G.; Mihm, M.C.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic Value of Tumor Infiltrating Lymphocytes in the Vertical Growth Phase of Primary Cutaneous Melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Mackensen, A.; Ferradini, L.; Carcelain, G.; Triebel, F.; Faure, F.; Viel, S.; Hercend, T. Evidence for in Situ Amplification of Cytotoxic T-Lymphocytes with Antitumor Activity in a Human Regressive Melanoma. Cancer Res. 1993, 53, 3569–3573. [Google Scholar]
- Mahmoud, S.M.A.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.S.; Ellis, I.O.; Green, A.R. Tumor-Infiltrating CD8+ Lymphocytes Predict Clinical Outcome in Breast Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Gomez, S.; Arenas, A.; El-Ashry, D.; Schmidt, M.; Gehrmann, M.; Caldas, C. Improved Prognostic Classification of Breast Cancer Defined by Antagonistic Activation Patterns of Immune Response Pathway Modules. BMC Cancer 2010, 10, 604. [Google Scholar] [CrossRef] [Green Version]
- Dahlin, A.M.; Henriksson, M.L.; Van Guelpen, B.; Stenling, R.; Oberg, A.; Rutegård, J.; Palmqvist, R. Colorectal Cancer Prognosis Depends on T-Cell Infiltration and Molecular Characteristics of the Tumor. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc 2011, 24, 671–682. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagès, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In Situ Cytotoxic and Memory T Cells Predict Outcome in Patients with Early-Stage Colorectal Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef]
- Dieu-Nosjean, M.-C.; Antoine, M.; Danel, C.; Heudes, D.; Wislez, M.; Poulot, V.; Rabbe, N.; Laurans, L.; Tartour, E.; de Chaisemartin, L.; et al. Long-Term Survival for Patients with Non-Small-Cell Lung Cancer with Intratumoral Lymphoid Structures. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4410–4417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, O.; Ishii, G.; Kubota, K.; Murata, Y.; Naito, Y.; Mizuno, T.; Aokage, K.; Saijo, N.; Nishiwaki, Y.; Gemma, A.; et al. Predominant Infiltration of Macrophages and CD8(+) T Cells in Cancer Nests Is a Significant Predictor of Survival in Stage IV Nonsmall Cell Lung Cancer. Cancer 2008, 113, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Iyama, K.; Ogawa, M.; Yoshioka, H.; Sado, Y.; Oohashi, T.; Ninomiya, Y. Differential Tissular Expression and Localization of Type IV Collagen Alpha1(IV), Alpha2(IV), Alpha5(IV), and Alpha6(IV) Chains and Their MRNA in Normal Breast and in Benign and Malignant Breast Tumors. Lab. Investig. J. Tech. Methods Pathol. 1999, 79, 281–292. [Google Scholar]
- Wakabayashi, O.; Yamazaki, K.; Oizumi, S.; Hommura, F.; Kinoshita, I.; Ogura, S.; Dosaka-Akita, H.; Nishimura, M. CD4+ T Cells in Cancer Stroma, Not CD8+ T Cells in Cancer Cell Nests, Are Associated with Favorable Prognosis in Human Non-Small Cell Lung Cancers. Cancer Sci. 2003, 94, 1003–1009. [Google Scholar] [CrossRef]
- Koreishi, A.F.; Saenz, A.J.; Persky, D.O.; Cui, H.; Moskowitz, A.; Moskowitz, C.H.; Teruya-Feldstein, J. The Role of Cytotoxic and Regulatory T Cells in Relapsed/Refractory Hodgkin Lymphoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2010, 18, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzankov, A.; Meier, C.; Hirschmann, P.; Went, P.; Pileri, S.A.; Dirnhofer, S. Correlation of High Numbers of Intratumoral FOXP3+ Regulatory T Cells with Improved Survival in Germinal Center-like Diffuse Large B-Cell Lymphoma, Follicular Lymphoma and Classical Hodgkin’s Lymphoma. Haematologica 2008, 93, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; de Sauvage, F.J. Influence of Tumour Micro-Environment Heterogeneity on Therapeutic Response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune Checkpoint Inhibitors of PD-L1 as Cancer Therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- Wensveen, F.M.; Jelenčić, V.; Polić, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef]
- Allez, M.; Tieng, V.; Nakazawa, A.; Treton, X.; Pacault, V.; Dulphy, N.; Caillat-Zucman, S.; Paul, P.; Gornet, J.-M.; Douay, C.; et al. CD4+NKG2D+ T Cells in Crohn’s Disease Mediate Inflammatory and Cytotoxic Responses through MICA Interactions. Gastroenterology 2007, 132, 2346–2358. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of Ligands for the NKG2D Activating Receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [Green Version]
- Le Bert, N.; Gasser, S. Advances in NKG2D Ligand Recognition and Responses by NK Cells. Immunol. Cell Biol. 2014, 92, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Coronella, J.A.; Telleman, P.; Kingsbury, G.A.; Truong, T.D.; Hays, S.; Junghans, R.P. Evidence for an Antigen-Driven Humoral Immune Response in Medullary Ductal Breast Cancer. Cancer Res. 2001, 61, 7889–7899. [Google Scholar]
- Milne, K.; Köbel, M.; Kalloger, S.E.; Barnes, R.O.; Gao, D.; Gilks, C.B.; Watson, P.H.; Nelson, B.H. Systematic Analysis of Immune Infiltrates in High-Grade Serous Ovarian Cancer Reveals CD20, FoxP3 and TIA-1 as Positive Prognostic Factors. PLoS ONE 2009, 4, e6412. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Richter, G.; Schüler, T.; Ibe, S.; Cao, X.; Blankenstein, T. B Cells Inhibit Induction of T Cell-Dependent Tumor Immunity. Nat. Med. 1998, 4, 627–630. [Google Scholar] [CrossRef]
- Andreu, P.; Johansson, M.; Affara, N.I.; Pucci, F.; Tan, T.; Junankar, S.; Korets, L.; Lam, J.; Tawfik, D.; DeNardo, D.G.; et al. FcRgamma Activation Regulates Inflammation-Associated Squamous Carcinogenesis. Cancer Cell 2010, 17, 121–134. [Google Scholar] [CrossRef] [Green Version]
- de Visser, K.E.; Korets, L.V.; Coussens, L.M. De Novo Carcinogenesis Promoted by Chronic Inflammation Is B Lymphocyte Dependent. Cancer Cell 2005, 7, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Schioppa, T.; Moore, R.; Thompson, R.G.; Rosser, E.C.; Kulbe, H.; Nedospasov, S.; Mauri, C.; Coussens, L.M.; Balkwill, F.R. B Regulatory Cells and the Tumor-Promoting Actions of TNF-α during Squamous Carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 10662–10667. [Google Scholar] [CrossRef] [Green Version]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B Cells, Plasma Cells and Antibody Repertoires in the Tumour Microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Spits, H. The Biology of Innate Lymphoid Cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Simoni, Y.; Newell, E.W. Dissecting Human ILC Heterogeneity: More than Just Three Subsets. Immunology 2018, 153, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bal, S.M.; Golebski, K.; Spits, H. Plasticity of Innate Lymphoid Cell Subsets. Nat. Rev. Immunol. 2020, 20, 552–565. [Google Scholar] [CrossRef]
- Li, J.; Doty, A.L.; Tang, Y.; Berrie, D.; Iqbal, A.; Tan, S.A.; Clare-Salzler, M.J.; Wallet, S.M.; Glover, S.C. Enrichment of IL-17A+ IFN-Γ+ and IL-22+ IFN-Γ+ T Cell Subsets Is Associated with Reduction of NKp44+ ILC3s in the Terminal Ileum of Crohn’s Disease Patients. Clin. Exp. Immunol. 2017, 190, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Cohen, I. DNA Damage Talks to Inflammation. Cytokine Growth Factor Rev. 2017, 33, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shin, T.; Kawano, T.; Sato, H.; Kondo, E.; Toura, I.; Kaneko, Y.; Koseki, H.; Kanno, M.; Taniguchi, M. Requirement for Valpha14 NKT Cells in IL-12-Mediated Rejection of Tumors. Science 1997, 278, 1623–1626. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Nakayama, T.; Kamada, N.; Kaneko, Y.; Harada, M.; Ogura, N.; Akutsu, Y.; Motohashi, S.; Iizasa, T.; Endo, H.; et al. Antitumor Cytotoxicity Mediated by Ligand-Activated Human V Alpha24 NKT Cells. Cancer Res. 1999, 59, 5102–5105. [Google Scholar] [PubMed]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; Cretney, E.; Trapani, J.A.; Taniguchi, M.; Kawano, T.; Pelikan, S.B.; Crowe, N.Y.; Godfrey, D.I. Differential Tumor Surveillance by Natural Killer (NK) and NKT Cells. J. Exp. Med. 2000, 191, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, T.; Onodera, H.; Tsuruyama, T.; Mori, A.; Nagayama, S.; Hiai, H.; Imamura, M. Increased Intratumor Valpha24-Positive Natural Killer T Cells: A Prognostic Factor for Primary Colorectal Carcinomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 7322–7327. [Google Scholar] [CrossRef] [Green Version]
- Terabe, M.; Matsui, S.; Noben-Trauth, N.; Chen, H.; Watson, C.; Donaldson, D.D.; Carbone, D.P.; Paul, W.E.; Berzofsky, J.A. NKT Cell-Mediated Repression of Tumor Immunosurveillance by IL-13 and the IL-4R-STAT6 Pathway. Nat. Immunol. 2000, 1, 515–520. [Google Scholar] [CrossRef]
- Meredith, M.M.; Liu, K.; Darrasse-Jeze, G.; Kamphorst, A.O.; Schreiber, H.A.; Guermonprez, P.; Idoyaga, J.; Cheong, C.; Yao, K.-H.; Niec, R.E.; et al. Expression of the Zinc Finger Transcription Factor ZDC (Zbtb46, Btbd4) Defines the Classical Dendritic Cell Lineage. J. Exp. Med. 2012, 209, 1153–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trovato, R.; Fiore, A.; Sartori, S.; Canè, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; De Sanctis, F.; Poffe, O.; et al. Immunosuppression by Monocytic Myeloid-Derived Suppressor Cells in Patients with Pancreatic Ductal Carcinoma Is Orchestrated by STAT3. J. Immunother. Cancer 2019, 7, 255. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 610303. [Google Scholar] [CrossRef]
- McAndrew, N.; DeMichele, A. Neoadjuvant Chemotherapy Considerations in Triple-Negative Breast Cancer. J. Target. Ther. Cancer 2018, 7, 52–69. [Google Scholar]
- Gentles, A.J.; Hui, A.B.-Y.; Feng, W.; Azizi, A.; Nair, R.V.; Bouchard, G.; Knowles, D.A.; Yu, A.; Jeong, Y.; Bejnood, A.; et al. A Human Lung Tumor Microenvironment Interactome Identifies Clinically Relevant Cell-Type Cross-Talk. Genome Biol. 2020, 21, 107. [Google Scholar] [CrossRef]
- Sarode, P.; Schaefer, M.B.; Grimminger, F.; Seeger, W.; Savai, R. Macrophage and Tumor Cell Cross-Talk Is Fundamental for Lung Tumor Progression: We Need to Talk. Front. Oncol. 2020, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Pumariño, C.; Collado, M.; Herrera, M.; Peña, C. Tumor Microenvironment in Metastatic Colorectal Cancer: The Arbitrator in Patients’ Outcome. Cancers 2021, 13, 1130. [Google Scholar] [CrossRef]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between Cancer Cells and Tumor Associated Macrophages Is Required for Mesenchymal Circulating Tumor Cell-Mediated Colorectal Cancer Metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef]
- Mezzasoma, L.; Costanzi, E.; Scarpelli, P.; Talesa, V.N.; Bellezza, I. Extracellular Vesicles from Human Advanced-Stage Prostate Cancer Cells Modify the Inflammatory Response of Microenvironment-Residing Cells. Cancers 2019, 11, 1276. [Google Scholar] [CrossRef] [Green Version]
- Elia, A.R.; Caputo, S.; Bellone, M. Immune Checkpoint-Mediated Interactions Between Cancer and Immune Cells in Prostate Adenocarcinoma and Melanoma. Front. Immunol. 2018, 9, 1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, E.C.F.; Brown, M.P.; Gargett, T.; Ebert, L.M. The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy. Cells 2021, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, H.; Roncali, L.; Rousseau, A.; Chérel, M.; Delneste, Y.; Jeannin, P.; Hindré, F.; Garcion, E. Targeting Tumor Associated Macrophages to Overcome Conventional Treatment Resistance in Glioblastoma. Front. Pharmacol. 2020, 11, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the Extracellular Matrix in Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the Tumour Stroma to Improve Cancer Therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Hynes, R.O. The Extracellular Matrix: Not Just Pretty Fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Gehler, S.; Baldassarre, M.; Lad, Y.; Leight, J.L.; Wozniak, M.A.; Riching, K.M.; Eliceiri, K.W.; Weaver, V.M.; Calderwood, D.A.; Keely, P.J. Filamin A-Beta1 Integrin Complex Tunes Epithelial Cell Response to Matrix Tension. Mol. Biol. Cell 2009, 20, 3224–3238. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.I.; Mouw, J.K.; Weaver, V.M. Biomechanical Regulation of Cell Orientation and Fate. Oncogene 2008, 27, 6981–6993. [Google Scholar] [CrossRef] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional Homeostasis and the Malignant Phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The Extracellular Matrix: A Dynamic Niche in Cancer Progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Remodeling and Homeostasis of the Extracellular Matrix: Implications for Fibrotic Diseases and Cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Kass, L.; Erler, J.T.; Dembo, M.; Weaver, V.M. Mammary Epithelial Cell: Influence of Extracellular Matrix Composition and Organization during Development and Tumorigenesis. Int. J. Biochem. Cell Biol. 2007, 39, 1987–1994. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilan, N.; Elkin, M.; Vlodavsky, I. Regulation, Function and Clinical Significance of Heparanase in Cancer Metastasis and Angiogenesis. Int. J. Biochem. Cell Biol. 2006, 38, 2018–2039. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Xie, T. Stem Cell Niche: Structure and Function. Annu. Rev. Cell Dev. Biol. 2005, 21, 605–631. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, B.B.; Gehlot, P. Inflammation and Cancer: How Friendly Is the Relationship for Cancer Patients? Curr. Opin. Pharmacol. 2009, 9, 351–369. [Google Scholar] [CrossRef] [Green Version]
- Feagins, L.A.; Souza, R.F.; Spechler, S.J. Carcinogenesis in IBD: Potential Targets for the Prevention of Colorectal Cancer. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 297–305. [Google Scholar] [CrossRef]
- Kawanishi, S.; Hiraku, Y.; Pinlaor, S.; Ma, N. Oxidative and Nitrative DNA Damage in Animals and Patients with Inflammatory Diseases in Relation to Inflammation-Related Carcinogenesis. Biol. Chem. 2006, 387, 365–372. [Google Scholar] [CrossRef]
- Murata, M.; Thanan, R.; Ma, N.; Kawanishi, S. Role of Nitrative and Oxidative DNA Damage in Inflammation-Related Carcinogenesis. J. Biomed. Biotechnol. 2012, 2012, 623019. [Google Scholar] [CrossRef]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-Analysis: Colorectal and Small Bowel Cancer Risk in Patients with Crohn’s Disease. Aliment. Pharmacol. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The Risk of Colorectal Cancer in Ulcerative Colitis: A Meta-Analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Jess, T.; Loftus, E.V.; Velayos, F.S.; Harmsen, W.S.; Zinsmeister, A.R.; Smyrk, T.C.; Schleck, C.D.; Tremaine, W.J.; Melton, L.J.; Munkholm, P.; et al. Risk of Intestinal Cancer in Inflammatory Bowel Disease: A Population-Based Study from Olmsted County, Minnesota. Gastroenterology 2006, 130, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Vainio, H.; Boffetta, P. Mechanisms of the Combined Effect of Asbestos and Smoking in the Etiology of Lung Cancer. Scand. J. Work. Environ. Health 1994, 20, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kato, J.; Inoue, I.; Yoshimura, N.; Deguchi, H.; Mukoubayashi, C.; Oka, M.; Watanabe, M.; Enomoto, S.; Niwa, T.; et al. Cancer Development Based on Chronic Active Gastritis and Resulting Gastric Atrophy as Assessed by Serum Levels of Pepsinogen and Helicobacter Pylori Antibody Titer. Int. J. Cancer 2014, 134, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Krieger, J.N.; Riley, D.E.; Vesella, R.L.; Miner, D.C.; Ross, S.O.; Lange, P.H. Bacterial Dna Sequences in Prostate Tissue from Patients with Prostate Cancer and Chronic Prostatitis. J. Urol. 2000, 164, 1221–1228. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of Viral Hepatitis and Hepatocellular Carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araldi, R.P.; Sant’Ana, T.A.; Módolo, D.G.; de Melo, T.C.; Spadacci-Morena, D.D.; de Cassia Stocco, R.; Cerutti, J.M.; de Souza, E.B. The Human Papillomavirus (HPV)-Related Cancer Biology: An Overview. Biomed. Pharmacother. 2018, 106, 1537–1556. [Google Scholar] [CrossRef] [Green Version]
- Lekakos, L.; Karidis, N.P.; Dimitroulis, D.; Tsigris, C.; Kouraklis, G.; Nikiteas, N. Barrett’s Esophagus with High-Grade Dysplasia: Focus on Current Treatment Options. World J. Gastroenterol. 2011, 17, 4174–4183. [Google Scholar] [CrossRef] [PubMed]
- Zabron, A.; Edwards, R.J.; Khan, S.A. The Challenge of Cholangiocarcinoma: Dissecting the Molecular Mechanisms of an Insidious Cancer. Dis. Model. Mech. 2013, 6, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Gutman, M.; Reich, R.; Bar-Eli, M. Ultraviolet B Irradiation Promotes Tumorigenic and Metastatic Properties in Primary Cutaneous Melanoma via Induction of Interleukin 8. Cancer Res. 1995, 55, 3669–3674. [Google Scholar] [CrossRef] [Green Version]
- Bats, A.S.; Zafrani, Y.; Pautier, P.; Duvillard, P.; Morice, P. Malignant Transformation of Abdominal Wall Endometriosis to Clear Cell Carcinoma: Case Report and Review of the Literature. Fertil. Steril. 2008, 90, 1197.e13–1197.e16. [Google Scholar] [CrossRef]
- Levin, B. Gallbladder Carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1999, 10 (Suppl. 4), 129–130. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and Chemokines: At the Crossroads of Cell Signalling and Inflammatory Disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. The Paradox of Pro-Inflammatory Cytokines in Cancer. Cancer Metastasis Rev. 2006, 25, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-Related Inflammation, the Seventh Hallmark of Cancer: Links to Genetic Instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, M.; Fabri, M.; Krutzik, S.R.; Lee, D.J.; Vu, D.M.; Sieling, P.A.; Montoya, D.; Liu, P.T.; Modlin, R.L. Interleukin-1β Triggers the Differentiation of Macrophages with Enhanced Capacity to Present Mycobacterial Antigen to T Cells. Immunology 2014, 141, 174–180. [Google Scholar] [CrossRef]
- Apte, R.N.; Krelin, Y.; Song, X.; Dotan, S.; Recih, E.; Elkabets, M.; Carmi, Y.; Dvorkin, T.; White, R.M.; Gayvoronsky, L.; et al. Effects of Micro-Environment- and Malignant Cell-Derived Interleukin-1 in Carcinogenesis, Tumour Invasiveness and Tumour-Host Interactions. Eur. J. Cancer Oxf. Engl. 1990 2006, 42, 751–759. [Google Scholar] [CrossRef]
- Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 9. [Google Scholar] [CrossRef]
- Voronov, E.; Apte, R.N. IL-1 in Colon Inflammation, Colon Carcinogenesis and Invasiveness of Colon Cancer. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2015, 8, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Lee, O.-Y.; Park, Y.; Seo, M.W.; Lee, D.-S. IL-1β Induces IL-6 Production and Increases Invasiveness and Estrogen-Independent Growth in a TG2-Dependent Manner in Human Breast Cancer Cells. BMC Cancer 2016, 16, 724. [Google Scholar] [CrossRef] [Green Version]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef]
- Akagi, Y.; Liu, W.; Xie, K.; Zebrowski, B.; Shaheen, R.M.; Ellis, L.M. Regulation of Vascular Endothelial Growth Factor Expression in Human Colon Cancer by Interleukin-1beta. Br. J. Cancer 1999, 80, 1506–1511. [Google Scholar] [CrossRef] [Green Version]
- Carmi, Y.; Dotan, S.; Rider, P.; Kaplanov, I.; White, M.R.; Baron, R.; Abutbul, S.; Huszar, M.; Dinarello, C.A.; Apte, R.N.; et al. The Role of IL-1β in the Early Tumor Cell-Induced Angiogenic Response. J. Immunol. 2013, 190, 3500–3509. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.M.; Varghese, S.; Xu, H.; Alexander, H.R. Interleukin-1 and Cancer Progression: The Emerging Role of Interleukin-1 Receptor Antagonist as a Novel Therapeutic Agent in Cancer Treatment. J. Transl. Med. 2006, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 Signaling as a Promising Target to Improve the Efficacy of Cancer Immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of Interleukin-6 in Cancer Progression and Therapeutic Resistance. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Rossi, J.-F.; Lu, Z.-Y.; Jourdan, M.; Klein, B. Interleukin-6 as a Therapeutic Target. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1248–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 Released by Colon Cancer-Associated Fibroblasts Is Critical for Tumour Angiogenesis: Anti-Interleukin-6 Receptor Antibody Suppressed Angiogenesis and Inhibited Tumour-Stroma Interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Fakhoury, M.; Negrulj, R.; Mooranian, A.; Al-Salami, H. Inflammatory Bowel Disease: Clinical Aspects and Treatments. J. Inflamm. Res. 2014, 7, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Pereira, R.; Faria, R.; Lago, P.; Torres, T. Infection and Malignancy Risk in Patients Treated with TNF Inhibitors for Immune-Mediated Inflammatory Diseases. Curr. Drug Saf. 2017, 12, 162–170. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waugh, D.J.J.; Wilson, C. The Interleukin-8 Pathway in Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikfalvi, A.; Billottet, C. The CC and CXC Chemokines: Major Regulators of Tumor Progression and the Tumor Microenvironment. Am. J. Physiol. Cell Physiol. 2020, 318, C542–C554. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kuang, Z.; Zhang, D.; Gao, Y.; Ying, M.; Wang, T. Reactive Oxygen Species in Immune Cells: A New Antitumor Target. Biomed. Pharmacother. 2021, 133, 110978. [Google Scholar] [CrossRef]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the Interleukin-10 Receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef]
- Zhu, L.; Shi, T.; Zhong, C.; Wang, Y.; Chang, M.; Liu, X. IL-10 and IL-10 Receptor Mutations in Very Early Onset Inflammatory Bowel Disease. Gastroenterol. Res. 2017, 10, 65–69. [Google Scholar] [CrossRef]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-Wide Meta-Analysis Increases to 71 the Number of Confirmed Crohn’s Disease Susceptibility Loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 Receptor Signaling in Innate Immune Cells Regulates Mucosal Immune Tolerance and Anti-Inflammatory Macrophage Function. Immunity 2014, 40, 706–719. [Google Scholar] [CrossRef] [Green Version]
- Coperchini, F.; Chiovato, L.; Ricci, G.; Croce, L.; Magri, F.; Rotondi, M. The Cytokine Storm in COVID-19: Further Advances in Our Understanding the Role of Specific Chemokines Involved. Cytokine Growth Factor Rev. 2021, 58, 82–91. [Google Scholar] [CrossRef]
- Rossi, J.-F.; Lu, Z.Y.; Massart, C.; Levon, K. Dynamic Immune/Inflammation Precision Medicine: The Good and the Bad Inflammation in Infection and Cancer. Front. Immunol. 2021, 12, 595722. [Google Scholar] [CrossRef] [PubMed]
- Cobb, D.A.; Lee, D.W. Cytokine Release Syndrome Biology and Management. Cancer J. Sudbury Mass 2021, 27, 119–125. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master Switch from Tumor-Promoting Inflammation to Antitumor Immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ Biology in Cancer Progression and Immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Coricello, A.; Mesiti, F.; Lupia, A.; Maruca, A.; Alcaro, S. Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates. Mol. Basel Switz. 2020, 25, 3321. [Google Scholar] [CrossRef]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A Multifaceted Oncoprotein. Growth Factors Chur Switz. 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory Regulatory Network Mediated by the Joint Action of NF-KB, STAT3, and AP-1 Factors Is Involved in Many Human Cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.-Q.; Du, W.-L.; Cai, M.-H.; Yao, J.-Y.; Zhao, Y.-Y.; Mou, X.-Z. The Roles of Tumor-Associated Macrophages in Tumor Angiogenesis and Metastasis. Cell. Immunol. 2020, 353, 104119. [Google Scholar] [CrossRef]
- Leng, K.; Xu, Y.; Kang, P.; Qin, W.; Cai, H.; Wang, H.; Ji, D.; Jiang, X.; Li, J.; Li, Z.; et al. Akirin2 Is Modulated by MiR-490-3p and Facilitates Angiogenesis in Cholangiocarcinoma through the IL-6/STAT3/VEGFA Signaling Pathway. Cell Death Dis. 2019, 10, 262. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Zhang, S.; Zhuang, Y.; Zhang, H.; Bai, J.; Hou, Q. Interleukin-17 Stimulates STAT3-Mediated Endothelial Cell Activation for Neutrophil Recruitment. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 2340–2356. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Q.; Zhang, Z.; Jiang, F.; Meng, X.; Yan, H. Interleukin-10 Overexpression Improves the Function of Endothelial Progenitor Cells Stimulated with TNF-α through the Activation of the STAT3 Signaling Pathway. Int. J. Mol. Med. 2015, 35, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF Drives Cancer-Initiating Stem Cells through VEGFR-2/Stat3 Signaling to Upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef]
- Chen, L.; Han, X. Anti-PD-1/PD-L1 Therapy of Human Cancer: Past, Present, and Future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef] [Green Version]
- Bloom, M.J.; Saksena, S.D.; Swain, G.P.; Behar, M.S.; Yankeelov, T.E.; Sorace, A.G. The Effects of IKK-Beta Inhibition on Early NF-Kappa-B Activation and Transcription of Downstream Genes. Cell. Signal. 2019, 55, 17–25. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta Links Inflammation and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.-W. NF-KappaB in Cancer: From Innocent Bystander to Major Culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Nuclear Factor-KappaB: The Enemy Within. Cancer Cell 2004, 6, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Lalle, G.; Twardowski, J.; Grinberg-Bleyer, Y. NF-ΚB in Cancer Immunity: Friend or Foe? Cells 2021, 10, 355. [Google Scholar] [CrossRef]
- Fan, Y.; Mao, R.; Yang, J. NF-ΚB and STAT3 Signaling Pathways Collaboratively Link Inflammation to Cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Papila, K.B.; Sozer, V.; Cigdem, K.P.; Durmus, S.; Kurtulus, D.; Papila, C.; Gelisgen, R.; Uzun, H. Circulating Nuclear Factor-Kappa B Mediates Cancer-Associated Inflammation in Human Breast and Colon Cancer. J. Med. Biochem. 2021, 40, 150–159. [Google Scholar] [CrossRef]
- Dong, F.; Zhou, X.; Li, C.; Yan, S.; Deng, X.; Cao, Z.; Li, L.; Tang, B.; Allen, T.D.; Liu, J. Dihydroartemisinin Targets VEGFR2 via the NF-ΚB Pathway in Endothelial Cells to Inhibit Angiogenesis. Cancer Biol. Ther. 2014, 15, 1479–1488. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Galisteo, R.; Gutkind, J.S. CXCL8/IL8 Stimulates Vascular Endothelial Growth Factor (VEGF) Expression and the Autocrine Activation of VEGFR2 in Endothelial Cells by Activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) Complex. J. Biol. Chem. 2009, 284, 6038–6042. [Google Scholar] [CrossRef] [Green Version]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting Nuclear Factor-Kappa B to Overcome Resistance to Chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- De, D.; Chowdhury, P.; Panda, S.K.; Ghosh, U. Ethanolic Extract of Leaf of Dillenia Pentagyna Reduces In-Vitro Cell Migration and Induces Intrinsic Pathway of Apoptosis via Downregulation of NF-Κβ in Human NSCLC A549 Cells. J. Cell. Biochem. 2019, 120, 19841–19857. [Google Scholar] [CrossRef]
- Huang, L.; Jian, Z.; Gao, Y.; Zhou, P.; Zhang, G.; Jiang, B.; Lv, Y. RPN2 Promotes Metastasis of Hepatocellular Carcinoma Cell and Inhibits Autophagy via STAT3 and NF-ΚB Pathways. Aging 2019, 11, 6674–6690. [Google Scholar] [CrossRef]
- Pu, D.; Yin, L.; Huang, L.; Qin, C.; Zhou, Y.; Wu, Q.; Li, Y.; Zhou, Q.; Li, L. Cyclooxygenase-2 Inhibitor: A Potential Combination Strategy With Immunotherapy in Cancer. Front. Oncol. 2021, 11, 637504. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and Cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Busquets-Cortés, C.; Capó, X.; Tejada, S.; Tur, J.A.; Pons, A.; Sureda, A. Cyclooxygenase-2 Inhibitors as a Therapeutic Target in Inflammatory Diseases. Curr. Med. Chem. 2019, 26, 3225–3241. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Bao, L.W.; Merajver, S.D. Tetrathiomolybdate Inhibits Angiogenesis and Metastasis through Suppression of the NFkappaB Signaling Cascade. Mol. Cancer Res. MCR 2003, 1, 701–706. [Google Scholar]
- Wang, D.; DuBois, R.N. Immunosuppression Associated with Chronic Inflammation in the Tumor Microenvironment. Carcinogenesis 2015, 36, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escuin-Ordinas, H.; Atefi, M.; Fu, Y.; Cass, A.; Ng, C.; Huang, R.R.; Yashar, S.; Comin-Anduix, B.; Avramis, E.; Cochran, A.J.; et al. COX-2 Inhibition Prevents the Appearance of Cutaneous Squamous Cell Carcinomas Accelerated by BRAF Inhibitors. Mol. Oncol. 2014, 8, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Panza, E.; De Cicco, P.; Ercolano, G.; Armogida, C.; Scognamiglio, G.; Anniciello, A.M.; Botti, G.; Cirino, G.; Ianaro, A. Differential Expression of Cyclooxygenase-2 in Metastatic Melanoma Affects Progression Free Survival. Oncotarget 2016, 7, 57077–57085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, J.; Lu, X.; Hu, Y.; Piao, C.; Wu, X.; Liu, X.; Huang, C.; Wang, Y.; Li, D.; Liu, J. Prostaglandin E2 and PD-1 Mediated Inhibition of Antitumor CTL Responses in the Human Tumor Microenvironment. Oncotarget 2017, 8, 89802–89810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, K.J.; Li, G. An Overview of Cancer Prevention: Chemoprevention and Immunoprevention. J. Cancer Prev. 2020, 25, 127–135. [Google Scholar] [CrossRef]
- Markosyan, N.; Li, J.; Sun, Y.H.; Richman, L.P.; Lin, J.H.; Yan, F.; Quinones, L.; Sela, Y.; Yamazoe, T.; Gordon, N.; et al. Tumor Cell-Intrinsic EPHA2 Suppresses Anti-Tumor Immunity by Regulating PTGS2 (COX-2). J. Clin. Investig. 2019, 129, 3594–3609. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.; Zhao, H.; Zhang, Q.; Zhou, Y.; Wu, L.; Lei, J.; Wang, X.; Zhang, J.; Zhang, X.; Zheng, L.; et al. A RIPK3-PGE2 Circuit Mediates Myeloid-Derived Suppressor Cell-Potentiated Colorectal Carcinogenesis. Cancer Res. 2018, 78, 5586–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Chen, P.; Leng, Y.; Kang, J. Histone Deacetylase 6 Regulates the Immunosuppressive Properties of Cancer-Associated Fibroblasts in Breast Cancer through the STAT3-COX2-Dependent Pathway. Oncogene 2018, 37, 5952–5966. [Google Scholar] [CrossRef]
- Chiang, K.-H.; Shieh, J.-M.; Shen, C.-J.; Chang, T.-W.; Wu, P.-T.; Hsu, J.-Y.; Tsai, J.-P.; Chang, W.-C.; Chen, B.-K. Epidermal Growth Factor-Induced COX-2 Regulates Metastasis of Head and Neck Squamous Cell Carcinoma through Upregulation of Angiopoietin-like 4. Cancer Sci. 2020, 111, 2004–2015. [Google Scholar] [CrossRef] [PubMed]
- Smakman, N.; Kranenburg, O.; Vogten, J.M.; Bloemendaal, A.L.A.; van Diest, P.; Borel Rinkes, I.H.M. Cyclooxygenase-2 Is a Target of KRASD12, Which Facilitates the Outgrowth of Murine C26 Colorectal Liver Metastases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 41–48. [Google Scholar]
- Lim, H.J.; Park, J.H.; Jo, C.; Yoon, K.; Koh, Y.H. Cigarette Smoke Extracts and Cadmium Induce COX-2 Expression through γ-Secretase-Mediated P38 MAPK Activation in C6 Astroglia Cells. PLoS ONE 2019, 14, e0212749. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Wang, Y.; Hemmings, B.A.; Rüegg, C.; Xue, G. PKB/Akt-Dependent Regulation of Inflammation in Cancer. Semin. Cancer Biol. 2018, 48, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Noorolyai, S.; Shajari, N.; Baghbani, E.; Sadreddini, S.; Baradaran, B. The Relation between PI3K/AKT Signalling Pathway and Cancer. Gene 2019, 698, 120–128. [Google Scholar] [CrossRef]
- Xu, K.; Liu, P.; Wei, W. MTOR Signaling in Tumorigenesis. Biochim. Biophys. Acta 2014, 1846, 638–654. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Massi, D.; Hemmings, B.A.; Mandalà, M.; Hu, Z.; Wicki, A.; Xue, G. AKT-Ions with a TWIST between EMT and MET. Oncotarget 2016, 7, 62767–62777. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Kijlstra, A.; Chen, Y.; Yang, P. IL-17A Stimulates the Production of Inflammatory Mediators via Erk1/2, P38 MAPK, PI3K/Akt, and NF-ΚB Pathways in ARPE-19 Cells. Mol. Vis. 2011, 17, 3072–3077. [Google Scholar]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.-Y.; Baldwin, A.S. Akt-Dependent Regulation of NF-{kappa}B Is Controlled by MTOR and Raptor in Association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Factor, V.; Oliver, A.L.; Panta, G.R.; Thorgeirsson, S.S.; Sonenshein, G.E.; Arsura, M. Roles of Akt/PKB and IKK Complex in Constitutive Induction of NF-KappaB in Hepatocellular Carcinomas of Transforming Growth Factor Alpha/c-Myc Transgenic Mice. Hepatol. Baltim. Md 2001, 34, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.-M.; Li, Q.-L.; Gao, Q.; Jiang, J.-H.; Zhu, K.; Huang, X.-Y.; Pan, J.-F.; Yan, J.; Hu, J.-H.; Wang, Z.; et al. IL-17 Induces AKT-Dependent IL-6/JAK2/STAT3 Activation and Tumor Progression in Hepatocellular Carcinoma. Mol. Cancer 2011, 10, 150. [Google Scholar] [CrossRef] [Green Version]
- Xue, G.; Zippelius, A.; Wicki, A.; Mandalà, M.; Tang, F.; Massi, D.; Hemmings, B.A. Integrated Akt/PKB Signaling in Immunomodulation and Its Potential Role in Cancer Immunotherapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.-Z.; Hu, X.-W.; Xia, C.; He, J.; Zhou, Q.; Shi, X.; Fang, J.; Jiang, B.-H. Reactive Oxygen Species Regulate Epidermal Growth Factor-Induced Vascular Endothelial Growth Factor and Hypoxia-Inducible Factor-1alpha Expression through Activation of AKT and P70S6K1 in Human Ovarian Cancer Cells. Free Radic. Biol. Med. 2006, 41, 1521–1533. [Google Scholar] [CrossRef]
- Madge, L.A.; Pober, J.S. A Phosphatidylinositol 3-Kinase/Akt Pathway, Activated by Tumor Necrosis Factor or Interleukin-1, Inhibits Apoptosis but Does Not Activate NFkappaB in Human Endothelial Cells. J. Biol. Chem. 2000, 275, 15458–15465. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Shen, H.; Xu, M.; Liu, O.; Zhao, L.; Liu, S.; Guo, Z.; Du, J. FRP Inhibits Ox-LDL-Induced Endothelial Cell Apoptosis through an Akt-NF-{kappa}B-Bcl-2 Pathway and Inhibits Endothelial Cell Apoptosis in an ApoE-Knockout Mouse Model. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E351–E363. [Google Scholar] [CrossRef]
- Massacesi, C.; Di Tomaso, E.; Urban, P.; Germa, C.; Quadt, C.; Trandafir, L.; Aimone, P.; Fretault, N.; Dharan, B.; Tavorath, R.; et al. PI3K Inhibitors as New Cancer Therapeutics: Implications for Clinical Trial Design. OncoTargets Ther. 2016, 9, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvarna, V.; Murahari, M.; Khan, T.; Chaubey, P.; Sangave, P. Phytochemicals and PI3K Inhibitors in Cancer-An Insight. Front. Pharmacol. 2017, 8, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef]
- Niehrs, C. The Complex World of WNT Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Azbazdar, Y.; Karabicici, M.; Erdal, E.; Ozhan, G. Regulation of Wnt Signaling Pathways at the Plasma Membrane and Their Misregulation in Cancer. Front. Cell Dev. Biol. 2021, 9, 631623. [Google Scholar] [CrossRef] [PubMed]
- Caspi, M.; Wittenstein, A.; Kazelnik, M.; Shor-Nareznoy, Y.; Rosin-Arbesfeld, R. Therapeutic Targeting of the Oncogenic Wnt Signaling Pathway for Treating Colorectal Cancer and Other Colonic Disorders. Adv. Drug Deliv. Rev. 2021, 169, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ortiz, M.A.; Kotula, L. The Physiological Role of Wnt Pathway in Normal Development and Cancer. Exp. Biol. Med. Maywood NJ 2020, 245, 411–426. [Google Scholar] [CrossRef]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and Mechanisms of WNT Pathway Tumour Suppressors in Cancer. Nat. Rev. Cancer 2021, 21, 5–21. [Google Scholar] [CrossRef]
- Rasola, A.; Fassetta, M.; De Bacco, F.; D’Alessandro, L.; Gramaglia, D.; Di Renzo, M.F.; Comoglio, P.M. A Positive Feedback Loop between Hepatocyte Growth Factor Receptor and Beta-Catenin Sustains Colorectal Cancer Cell Invasive Growth. Oncogene 2007, 26, 1078–1087. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.; Bui, T.D.; Poulsom, R.; Kaklamanis, L.; Williams, G.; Harris, A.L. Up-Regulation of Macrophage Wnt Gene Expression in Adenoma-Carcinoma Progression of Human Colorectal Cancer. Br. J. Cancer 1999, 81, 496–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojalvo, L.S.; Whittaker, C.A.; Condeelis, J.S.; Pollard, J.W. Gene Expression Analysis of Macrophages That Facilitate Tumor Invasion Supports a Role for Wnt-Signaling in Mediating Their Activity in Primary Mammary Tumors. J. Immunol. 2010, 184, 702–712. [Google Scholar] [CrossRef] [Green Version]
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 Promotes Colon Cancer Cell Growth through a Gs-Axin-Beta-Catenin Signaling Axis. Science 2005, 310, 1504–1510. [Google Scholar] [CrossRef]
- Lopez-Bergami, P.; Barbero, G. The Emerging Role of Wnt5a in the Promotion of a Pro-Inflammatory and Immunosuppressive Tumor Microenvironment. Cancer Metastasis Rev. 2020, 39, 933–952. [Google Scholar] [CrossRef] [PubMed]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, C.; Schaer, D.J.; Bachli, E.B.; Kurrer, M.O.; Schoedon, G. Wnt5A/CaMKII Signaling Contributes to the Inflammatory Response of Macrophages and Is a Target for the Antiinflammatory Action of Activated Protein C and Interleukin-10. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 504–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.-P.; Gan, Y.-H.; Zhang, C.-G.; Zhou, C.-Y.; Ma, K.-T.; Meng, J.-H.; Ma, X.-C. Requirement of the NF-ΚB Pathway for Induction of Wnt-5A by Interleukin-1β in Condylar Chondrocytes of the Temporomandibular Joint: Functional Crosstalk between the Wnt-5A and NF-ΚB Signaling Pathways. Osteoarthr. Cartil. 2011, 19, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Pérez-Hernández, A.I.; Gurbindo, J.; Ramírez, B.; Méndez-Giménez, L.; Rotellar, F.; Valentí, V.; Moncada, R.; et al. Activation of Noncanonical Wnt Signaling through WNT5A in Visceral Adipose Tissue of Obese Subjects Is Related to Inflammation. J. Clin. Endocrinol. Metab. 2014, 99, E1407–E1417. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Kang, M.-J.; Han, J.-S. Interleukin-1 Beta Promotes Neuronal Differentiation through the Wnt5a/RhoA/JNK Pathway in Cortical Neural Precursor Cells. Mol. Brain 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Linnskog, R.; Mohapatra, P.; Moradi, F.; Prasad, C.P.; Andersson, T. Demonstration of a WNT5A-IL-6 Positive Feedback Loop in Melanoma Cells: Dual Interference of This Loop More Effectively Impairs Melanoma Cell Invasion. Oncotarget 2016, 7, 37790–37802. [Google Scholar] [CrossRef] [Green Version]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Jain, P.; Ferrajoli, A.; Burger, J.A.; Bose, P.; Thompson, P.A.; Jain, N.; et al. STAT3-Induced Wnt5a Provides Chronic Lymphocytic Leukemia Cells with Survival Advantage. J. Immunol. 2019, 203, 3078–3085. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, C.; Wang, S.; Shi, D.; Wei, C.; Song, J.; Lin, X.; Dou, R.; Bai, J.; Xiang, Z.; et al. Wnt5a-Induced M2 Polarization of Tumor-Associated Macrophages via IL-10 Promotes Colorectal Cancer Progression. Cell Commun. Signal. CCS 2020, 18, 51. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, L.; Yu, J.; Ghia, E.M.; Choi, M.Y.; Zhang, L.; Zhang, S.; Sanchez-Lopez, E.; Widhopf, G.F.; Messer, K.; et al. Cirmtuzumab Blocks Wnt5a/ROR1 Stimulation of NF-ΚB to Repress Autocrine STAT3 Activation in Chronic Lymphocytic Leukemia. Blood 2019, 134, 1084–1094. [Google Scholar] [CrossRef]
- Barbero, G.; Castro, M.V.; Villanueva, M.B.; Quezada, M.J.; Fernández, N.B.; DeMorrow, S.; Lopez-Bergami, P. An Autocrine Wnt5a Loop Promotes NF-ΚB Pathway Activation and Cytokine/Chemokine Secretion in Melanoma. Cells 2019, 8, 1060. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Nishita, M.; Hoshi, K.; Honda, T.; Kakeji, Y.; Minami, Y. Mesenchymal Stem Cell-Derived CXCL16 Promotes Progression of Gastric Cancer Cells by STAT3-Mediated Expression of Ror1. Cancer Sci. 2020, 111, 1254–1265. [Google Scholar] [CrossRef] [Green Version]
- Bissell, M.J.; Hines, W.C. Why Don’t We Get More Cancer? A Proposed Role of the Microenvironment in Restraining Cancer Progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Butti, R.; Nimma, R.; Kundu, G.; Bulbule, A.; Kumar, T.V.S.; Gunasekaran, V.P.; Tomar, D.; Kumar, D.; Mane, A.; Gill, S.S.; et al. Tumor-Derived Osteopontin Drives the Resident Fibroblast to Myofibroblast Differentiation through Twist1 to Promote Breast Cancer Progression. Oncogene 2021, 40, 2002–2017. [Google Scholar] [CrossRef]
- Wei, Y.; Xiao, X.; Lao, X.-M.; Zheng, L.; Kuang, D.-M. Immune Landscape and Therapeutic Strategies: New Insights into PD-L1 in Tumors. Cell. Mol. Life Sci. CMLS 2021, 78, 867–887. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Greten, F.R. The Inflammatory Pathogenesis of Colorectal Cancer. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef]
- Rhodes, J.M.; Campbell, B.J. Inflammation and Colorectal Cancer: IBD-Associated and Sporadic Cancer Compared. Trends Mol. Med. 2002, 8, 10–16. [Google Scholar] [CrossRef]
- De Pergola, G.; Silvestris, F. Obesity as a Major Risk Factor for Cancer. J. Obes. 2013, 2013, 291546. [Google Scholar] [CrossRef] [Green Version]
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the Malignant Phenotype of Human Breast Cells in Three-Dimensional Culture and in Vivo by Integrin Blocking Antibodies. J. Cell Biol. 1997, 137, 231–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Swain, C.A.; Shevde, L.A. Informing the New Developments and Future of Cancer Immunotherapy: Future of Cancer Immunotherapy. Cancer Metastasis Rev. 2021. [Google Scholar] [CrossRef]
- Zhong, S.; Jeong, J.-H.; Chen, Z.; Chen, Z.; Luo, J.-L. Targeting Tumor Microenvironment by Small-Molecule Inhibitors. Transl. Oncol. 2020, 13, 57–69. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural Basis for Small Molecule Targeting of the Programmed Death Ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [Green Version]
- Zollo, M.; Di Dato, V.; Spano, D.; De Martino, D.; Liguori, L.; Marino, N.; Vastolo, V.; Navas, L.; Garrone, B.; Mangano, G.; et al. Targeting Monocyte Chemotactic Protein-1 Synthesis with Bindarit Induces Tumor Regression in Prostate and Breast Cancer Animal Models. Clin. Exp. Metastasis 2012, 29, 585–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grither, W.R.; Longmore, G.D. Inhibition of Tumor-Microenvironment Interaction and Tumor Invasion by Small-Molecule Allosteric Inhibitor of DDR2 Extracellular Domain. Proc. Natl. Acad. Sci. USA 2018, 115, E7786–E7794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic Antibodies: Successes, Limitations and Hopes for the Future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, N.; Popel, A.S. Peptides That Immunoactivate the Tumor Microenvironment. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188486. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, J.F.S.; Jordan, P.; Matos, P. A Signaling View into the Inflammatory Tumor Microenvironment. Immuno 2021, 1, 91-118. https://doi.org/10.3390/immuno1020007
Pereira JFS, Jordan P, Matos P. A Signaling View into the Inflammatory Tumor Microenvironment. Immuno. 2021; 1(2):91-118. https://doi.org/10.3390/immuno1020007
Chicago/Turabian StylePereira, Joana F. S., Peter Jordan, and Paulo Matos. 2021. "A Signaling View into the Inflammatory Tumor Microenvironment" Immuno 1, no. 2: 91-118. https://doi.org/10.3390/immuno1020007
APA StylePereira, J. F. S., Jordan, P., & Matos, P. (2021). A Signaling View into the Inflammatory Tumor Microenvironment. Immuno, 1(2), 91-118. https://doi.org/10.3390/immuno1020007