
CIDP: Current Treatments and Identification of Targets for Future Specific Therapeutic Intervention



Abstract
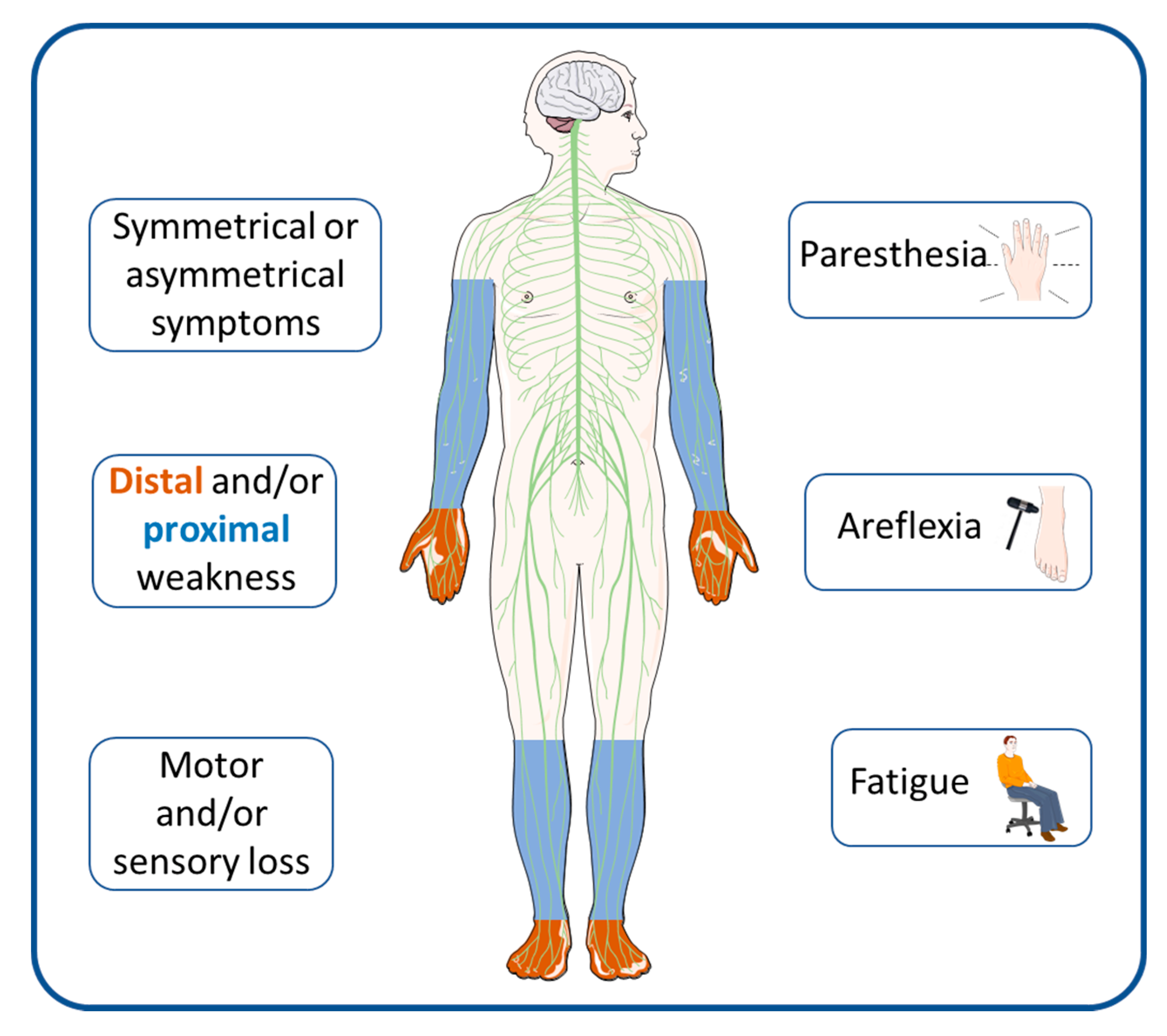
:1. Introduction
2. Prognosis
3. Current Treatments
4. Animal Models
5. Novel Therapeutic Options
5.1. IVIG and SCIg
5.2. B Cell-Targeted Strategies
5.3. T Cell-Targeted Strategies
5.4. Ig-Targeted Strategies
5.5. Complement-Targeted Strategies
5.6. Antigen-Presenting Cells (APCs) and Autophagy-Targeted Strategies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Eftimov, F.; van Schaik, I. Chronic inflammatory demyelinating polyradiculoneuropathy: Update on clinical features, phenotypes and treatment options. Curr. Opin. Neurol. 2013, 26, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Hafsteinsdottir, B.; Olafsson, E. Incidence and Natural History of Idiopathic Chronic Inflammatory Demyelinating Polyneuropathy: A Population-Based Study in Iceland. Eur. Neurol. 2016, 75, 263–268. [Google Scholar] [CrossRef]
- Mahdi-Rogers, M.; Hughes, R.A.C. Epidemiology of chronic inflammatory neuropathies in southeast England. Eur. J. Neurol. 2014, 21, 28–33. [Google Scholar] [CrossRef]
- Chiò, A.; Cocito, D.; Bottacchi, E.; Buffa, C.; Leone, M.; Plano, F.; Mutani, R.; Calvo, A.; Parcidp, T. Idiopathic chronic inflammatory demyelinating polyneuropathy: An epidemiological study in Italy. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1349–1353. [Google Scholar] [CrossRef] [Green Version]
- Mygland, A.; Monstad, P. Chronic polyneuropathies in Vest-Agder, Norway. Eur. J. Neurol. 2001, 8, 157–165. [Google Scholar] [CrossRef]
- Kusumi, M.; Nakashima, K.; Nakayama, H.; Takahashi, K. Epidemiology of inflammatory neurological and inflammatory neuromuscular diseases in Tottori Prefecture, Japan. Psychiatry Clin. Neurosci. 1995, 49, 169–174. [Google Scholar] [CrossRef]
- Laughlin, R.S.; Dyck, P.J.; Melton, L.J.; Leibson, C.; Ransom, J.; Dyck, P.J.B. Incidence and prevalence of CIDP and the association of diabetes mellitus. Neurology 2009, 73, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Lefter, S.; Hardiman, O.; Ryan, A.M. A population-based epidemiologic study of adult neuromuscular disease in the Republic of Ireland. Neurology 2017, 88, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, H.C.; Burke, D.; Kuwabara, S. Chronic inflammatory demyelinating polyneuropathy: Update on diagnosis, immunopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2019, 90, 981–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajabally, Y.A.; Sarasamma, P.; Abbott, R.J. Chronic inflammatory demyelinating polyneuropathy after Campylobacter jejuni infection mimicking vasculitic mononeuritis multiplex in a diabetic. J. Peripher. Nerv. Syst. JPNS 2004, 9, 98–103. [Google Scholar] [CrossRef]
- Meléndez-Vásquez, C.; Redford, J.; Choudhary, P.P.; Gray, I.A.; Maitland, P.; Gregson, N.A.; Smith, K.J.; Hughes, R.A. Immunological investigation of chronic inflammatory demyelinating polyradiculoneuropathy. J. Neuroimmunol. 1997, 73, 124–134. [Google Scholar] [CrossRef]
- Shahrizaila, N.; Lehmann, H.C.; Kuwabara, S. Guillain-Barré syndrome. Lancet 2021, 397, 1214–1228. [Google Scholar] [CrossRef]
- Rodríguez, Y.; Vatti, N.; Ramírez-Santana, C.; Chang, C.; Mancera-Páez, O.; Gershwin, M.E.; Anaya, J.-M. Chronic inflammatory demyelinating polyneuropathy as an autoimmune disease. J. Autoimmun. 2019, 102, 8–37. [Google Scholar] [CrossRef]
- Segal, Y.; Shoenfeld, Y. Vaccine-induced autoimmunity: The role of molecular mimicry and immune crossreaction. Cell. Mol. Immunol. 2018, 15, 586–594. [Google Scholar] [CrossRef]
- Yuki, N.; Taki, T.; Inagaki, F.; Kasama, T.; Takahashi, M.; Saito, K.; Handa, S.; Miyatake, T. A bacterium lipopolysaccharide that elicits Guillain-Barré syndrome has a GM1 ganglioside-like structure. J. Exp. Med. 1993, 178, 1771–1775. [Google Scholar] [CrossRef] [Green Version]
- Hafer-Macko, C.; Hsieh, S.T.; Li, C.Y.; Ho, T.W.; Sheikh, K.; Cornblath, D.R.; McKhann, G.M.; Asbury, A.K.; Griffin, J.W. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Ann. Neurol. 1996, 40, 635–644. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. Pathophysiology of Chronic Inflammatory Demyelinating Polyneuropathy: Insights into Classification and Therapeutic Strategy. Neurol. Ther. 2020, 9, 213–227. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. Macrophages and Autoantibodies in Demyelinating Diseases. Cells 2021, 10, 844. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Suzuki, S.; Takada, K.; Kusunoki, S. Antibodies to LM1 and LM1-containing ganglioside complexes in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J. Neuroimmunol. 2011, 239, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Vural, A.; Doppler, K.; Meinl, E. Autoantibodies Against the Node of Ranvier in Seropositive Chronic Inflammatory Demyelinating Polyneuropathy: Diagnostic, Pathogenic, and Therapeutic Relevance. Front. Immunol. 2018, 9, 1029. [Google Scholar] [CrossRef] [Green Version]
- Hagen, K.M.; Ousman, S.S. The immune response and aging in chronic inflammatory demyelinating polyradiculoneuropathy. J. Neuroinflamm. 2021, 18, 78. [Google Scholar] [CrossRef]
- Kuwabara, S.; Misawa, S.; Mori, M.; Tamura, N.; Kubota, M.; Hattori, T. Long term prognosis of chronic inflammatory demyelinating polyneuropathy: A five year follow up of 38 cases. J. Neurol. Neurosurg. Psychiatry 2006, 77, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Eftimov, F.; Vermeulen, M.; van Doorn, P.A.; Brusse, E.; van Schaik, I.N. Long-term remission of CIDP after pulsed dexamethasone or short-term prednisolone treatment. Neurology 2012, 78, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Vallat, J.-M.; Sommer, C.; Magy, L. Chronic inflammatory demyelinating polyradiculoneuropathy: Diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol. 2010, 9, 402–412. [Google Scholar] [CrossRef]
- Mygland, A.; Monstad, P.; Vedeler, C. Onset and course of chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 2005, 31, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Köller, H.; Kieseier, B.C.; Jander, S.; Hartung, H.-P. Chronic inflammatory demyelinating polyneuropathy. N. Engl. J. Med. 2005, 352, 1343–1356. [Google Scholar] [CrossRef] [Green Version]
- Dyck, P.J.; Lais, A.C.; Ohta, M.; Bastron, J.A.; Okazaki, H.; Groover, R.V. Chronic inflammatory polyradiculoneuropathy. Mayo Clin. Proc. 1975, 50, 621–637. [Google Scholar]
- Dyck, P.J.; O’Brien, P.C.; Oviatt, K.F.; Dinapoli, R.P.; Daube, J.R.; Bartleson, J.D.; Mokri, B.; Swift, T.; Low, P.A.; Windebank, A.J. Prednisone improves chronic inflammatory demyelinating polyradiculoneuropathy more than no treatment. Ann. Neurol. 1982, 11, 136–141. [Google Scholar] [CrossRef]
- Hughes, R.A.; Mehndiratta, M.M.; Rajabally, Y.A. Corticosteroids for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst. Rev. 2017, 11, CD002062. [Google Scholar] [CrossRef] [PubMed]
- Kivity, S.; Katz, U.; Daniel, N.; Nussinovitch, U.; Papageorgiou, N.; Shoenfeld, Y. Evidence for the use of intravenous immunoglobulins--a review of the literature. Clin. Rev. Allergy Immunol. 2010, 38, 201–269. [Google Scholar] [CrossRef]
- Chapman, J.; Shoenfeld, Y. Chronic inflammatory demyelinating polyradiculoneuropathy: Revisiting the role of intravenous immmunoglobulins. Isr. Med. Assoc. J. IMAJ 2013, 15, 293–294. [Google Scholar]
- Markvardsen, L.K.; Carstens, A.-K.R.; Knak, K.L.; Overgaard, K.; Vissing, J.; Andersen, H. Muscle Strength and Aerobic Capacity in Patients with CIDP One Year after Participation in an Exercise Trial. J. Neuromuscul. Dis. 2019, 6, 93–97. [Google Scholar] [CrossRef]
- Garssen, M.P.J.; Bussmann, J.B.J.; Schmitz, P.I.M.; Zandbergen, A.; Welter, T.G.; Merkies, I.S.J.; Stam, H.J.; van Doorn, P.A. Physical training and fatigue, fitness, and quality of life in Guillain-Barré syndrome and CIDP. Neurology 2004, 63, 2393–2395. [Google Scholar] [CrossRef]
- van Lieverloo, G.G.A.; Peric, S.; Doneddu, P.E.; Gallia, F.; Nikolic, A.; Wieske, L.; Verhamme, C.; Van Schaik, I.N.; Nobile-Orazio, E.; Basta, I.; et al. Corticosteroids in chronic inflammatory demyelinating polyneuropathy: A retrospective, multicentre study, comparing efficacy and safety of daily prednisolone, pulsed dexamethasone, and pulsed intravenous methylprednisolone. J. Neurol. 2018, 265, 2052–2059. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C.; Engel, W.K. Chronic relapsing (dysimmune) polyneuropathy: Pathogenesis and treatment. Ann. Neurol. 1981, 9, 134–145. [Google Scholar] [CrossRef]
- Nobile-Orazio, E.; Cocito, D.; Jann, S.; Uncini, A.; Beghi, E.; Messina, P.; Antonini, G.; Fazio, R.; Gallia, F.; Schenone, A.; et al. Intravenous immunoglobulin versus intravenous methylprednisolone for chronic inflammatory demyelinating polyradiculoneuropathy: A randomised controlled trial. Lancet Neurol. 2012, 11, 493–502. [Google Scholar] [CrossRef]
- Bus, S.R.M.; Zambreanu, L.; Abbas, A.; Rajabally, Y.A.; Hadden, R.D.M.; de Haan, R.J.; de Borgie, C.A.J.M.; Lunn, M.P.; van Schaik, I.N.; Eftimov, F.; et al. Intravenous immunoglobulin and intravenous methylprednisolone as optimal induction treatment in chronic inflammatory demyelinating polyradiculoneuropathy: Protocol of an international, randomised, double-blind, placebo-controlled trial (OPTIC). Trials 2021, 22, 155. [Google Scholar] [CrossRef] [PubMed]
- Adrichem, M.E.; Bus, S.R.; Wieske, L.; Mohammed, H.; Verhamme, C.; Hadden, R.; Van Schaik, I.N.; Eftimov, F. Combined intravenous immunoglobulin and methylprednisolone as induction treatment in chronic inflammatory demyelinating polyneuropathy (OPTIC protocol): A prospective pilot study. Eur. J. Neurol. 2020, 27, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Hughes, R.; Bensa, S.; Willison, H.; Van den Bergh, P.; Comi, G.; Illa, I.; Nobile-Orazio, E.; Van Doorn, P.; Dalakas, M.; Bojar, M.; et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann. Neurol. 2001, 50, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Mehndiratta, M.M.; Hughes, R.A.C.; Pritchard, J. Plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst. Rev. 2015, 2015, CD003906. [Google Scholar] [CrossRef]
- Hadden, R.D.M.; Bensa, S.; Lunn, M.P.T.; Hughes, R.A.C. Immunoadsorption inferior to plasma exchange in a patient with chronic inflammatory demyelinating polyradiculoneuropathy. J. Neurol. Neurosurg. Psychiatry 2002, 72, 644–646. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, H.C.; Hartung, H.-P. Plasma exchange and intravenous immunoglobulins: Mechanism of action in immune-mediated neuropathies. J. Neuroimmunol. 2011, 231, 61–69. [Google Scholar] [CrossRef]
- Arnson, Y.; Shoenfeld, Y.; Amital, H. Intravenous immunoglobulin therapy for autoimmune diseases. Autoimmunity 2009, 42, 553–560. [Google Scholar] [CrossRef]
- Hahn, A.F.; Bolton, C.F.; Zochodne, D.; Feasby, T.E. Intravenous immunoglobulin treatment in chronic inflammatory demyelinating polyneuropathy. A double-blind, placebo-controlled, cross-over study. Brain J. Neurol. 1996, 119 Pt 4, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Barohn, R.J.; Freimer, M.L.; Kissel, J.T.; King, W.; Nagaraja, H.N.; Rice, R.; Campbell, W.; Donofrio, P.; Jackson, C.; et al. Randomized controlled trial of IVIg in untreated chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 2001, 56, 445–449. [Google Scholar] [CrossRef]
- Eftimov, F.; Winer, J.B.; Vermeulen, M.; de Haan, R.; van Schaik, I.N. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst. Rev. 2013, 12, CD001797. [Google Scholar] [CrossRef]
- Hughes, R.A.C.; Donofrio, P.; Bril, V.; Dalakas, M.C.; Deng, C.; Hanna, K.; Hartung, H.-P.; Latov, N.; Merkies, I.S.; van Doorn, P.A. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): A randomised placebo-controlled trial. Lancet Neurol. 2008, 7, 136–144. [Google Scholar] [CrossRef]
- Shalem, D.; Shemer, A.; Shovman, O.; Shoenfeld, Y.; Kivity, S. The Efficacy of Intravenous Immunoglobulin in Guillain-Barré Syndrome: The Experience of a Tertiary Medical Center. Isr. Med. Assoc. J. IMAJ 2018, 20, 754–760. [Google Scholar]
- Harel, M.; Shoenfeld, Y. Intravenous immunoglobulin and Guillain-Barré syndrome. Clin. Rev. Allergy Immunol. 2005, 29, 281–287. [Google Scholar] [CrossRef]
- Debs, R.; Reach, P.; Cret, C.; Demeret, S.; Saheb, S.; Maisonobe, T.; Viala, K. A new treatment regimen with high-dose and fractioned immunoglobulin in a special subgroup of severe and dependent CIDP patients. Int. J. Neurosci. 2017, 127, 864–872. [Google Scholar] [CrossRef]
- Orbach, H.; Katz, U.; Sherer, Y.; Shoenfeld, Y. Intravenous immunoglobulin: Adverse effects and safe administration. Clin. Rev. Allergy Immunol. 2005, 29, 173–184. [Google Scholar] [CrossRef]
- van Schaik, I.N.; Bril, V.; van Geloven, N.; Hartung, H.-P.; Lewis, R.A.; Sobue, G.; Lawo, J.-P.; Praus, M.; Mielke, O.; Durn, B.L.; et al. Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy (PATH): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2018, 17, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Beydoun, S.R.; Sharma, K.R.; Bassam, B.A.; Pulley, M.T.; Shije, J.Z.; Kafal, A. Individualizing Therapy in CIDP: A Mini-Review Comparing the Pharmacokinetics of Ig with SCIg and IVIg. Front. Neurol. 2021, 12, 638816. [Google Scholar] [CrossRef]
- Gentile, L.; Mazzeo, A.; Russo, M.; Arimatea, I.; Vita, G.; Toscano, A. Long-term treatment with subcutaneous immunoglobulin in patients with chronic inflammatory demyelinating polyradiculoneuropathy: A follow-up period up to 7 years. Sci. Rep. 2020, 10, 7910. [Google Scholar] [CrossRef] [PubMed]
- Querol, L.; Nogales-Gadea, G.; Rojas-Garcia, R.; Martinez-Hernandez, E.; Diaz-Manera, J.; Suárez-Calvet, X.; Navas, M.; Araque, J.; Gallardo, E.; Illa, I. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann. Neurol. 2013, 73, 370–380. [Google Scholar] [CrossRef]
- Devaux, J.J.; Miura, Y.; Fukami, Y.; Inoue, T.; Manso, C.; Belghazi, M.; Sekiguchi, K.; Kokubun, N.; Ichikawa, H.; Wong, A.H.Y.; et al. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 2016, 86, 800–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, Y.; Devaux, J.J.; Fukami, Y.; Manso, C.; Belghazi, M.; Wong, A.H.Y.; Yuki, N. Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain J. Neurol. 2015, 138, 1484–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomelli, R.; Afeltra, A.; Bartoloni, E.; Berardicurti, O.; Bombardieri, M.; Bortoluzzi, A.; Carubbi, F.; Caso, F.; Cervera, R.; Ciccia, F.; et al. The growing role of precision medicine for the treatment of autoimmune diseases; results of a systematic review of literature and Experts’ Consensus. Autoimmun. Rev. 2021, 20, 102738. [Google Scholar] [CrossRef]
- Conrad, K.; Shoenfeld, Y.; Fritzler, M.J. Precision health: A pragmatic approach to understanding and addressing key factors in autoimmune diseases. Autoimmun. Rev. 2020, 19, 102508. [Google Scholar] [CrossRef]
- Allen, J.A.; Berger, M.; Querol, L.; Kuitwaard, K.; Hadden, R.D. Individualized immunoglobulin therapy in chronic immune-mediated peripheral neuropathies. J. Peripher. Nerv. Syst. JPNS 2018, 23, 78–87. [Google Scholar] [CrossRef]
- Schafflick, D.; Kieseier, B.C.; Wiendl, H.; Zu, M.; Horste, G. Novel pathomechanisms in inflammatory neuropathies. J. Neuroinflamm. 2017, 14, 232. [Google Scholar] [CrossRef] [Green Version]
- Waksman, B.H.; Adams, R.D. Allergic neuritis: An experimental disease of rabbits induced by the injection of peripheral nervous tissue and adjuvants. J. Exp. Med. 1955, 102, 213–236. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.P.; Ljunggren, H.G.; Levi, M.; Nennesmo, I.; Wahren, B.; Mix, E.; Winblad, B.; Schalling, M.; Zhu, J. P0 protein peptide 180–199 together with pertussis toxin induces experimental autoimmune neuritis in resistant C57BL/6 mice. J. Neurosci. Res. 2000, 62, 717–721. [Google Scholar] [CrossRef]
- Gonsalvez, D.G.; Yoo, S.; Craig, G.A.; Wood, R.J.; Fletcher, J.L.; Murray, S.S.; Xiao, J. Myelin Protein Zero180-199 Peptide Induced Experimental Autoimmune Neuritis in C57BL/6 Mice. Methods Mol. Biol. 2018, 1791, 243–250. [Google Scholar] [CrossRef]
- Calida, D.M.; Kremlev, S.G.; Fujioka, T.; Hilliard, B.; Ventura, E.; Constantinescu, C.S.; Lavi, E.; Rostami, A. Experimental allergic neuritis in the SJL/J mouse: Induction of severe and reproducible disease with bovine peripheral nerve myelin and pertussis toxin with or without interleukin-12. J. Neuroimmunol. 2000, 107, 247. [Google Scholar] [CrossRef]
- Yuan, X.-J.; Wei, Y.-J.; Ao, Q.; Gong, K.; Wang, J.-Y.; Sun, Q.-S.; Zhang, L.; Zheng, Z.-C.; Chen, L. Myelin ultrastructure of sciatic nerve in rat experimental autoimmune neuritis model and its correlation with associated protein expression. Int. J. Clin. Exp. Pathol. 2015, 8, 7849–7858. [Google Scholar]
- Sheremata, W.A.; Behan, P.O. Experimental allergic neuritis: A new experimental approach. J. Neurol. Neurosurg. Psychiatry 1973, 36, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspary, E.A.; Field, E.J. Antibody response to central and peripheral nerve antigens in rat and guinea-pig. J. Neurol. Neurosurg. Psychiatry 1965, 28, 179–182. [Google Scholar] [CrossRef] [Green Version]
- Snyder, D.H.; Stone, S.H.; Raine, C.S. Attempts to induce chronic experimental allergic neuritis in strain 13 and Hartley guinea pigs. J. Neuropathol. Exp. Neurol. 1977, 36, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Miletic, H.; Utermöhlen, O.; Wedekind, C.; Hermann, M.; Stenzel, W.; Lassmann, H.; Schlüter, D.; Deckert, M. P0(106–125) is a neuritogenic epitope of the peripheral myelin protein P0 and induces autoimmune neuritis in C57BL/6 mice. J. Neuropathol. Exp. Neurol. 2005, 64, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Brostoff, S.W.; Levit, S.; Powers, J.M. Induction of experimental allergic neuritis with a peptide from myelin P2 basic protein. Nature 1977, 268, 752–753. [Google Scholar] [CrossRef] [PubMed]
- Rostami, A.; Gregorian, S.K.; Brown, M.J.; Pleasure, D.E. Induction of severe experimental autoimmune neuritis with a synthetic peptide corresponding to the 53–78 amino acid sequence of the myelin P2 protein. J. Neuroimmunol. 1990, 30, 145–151. [Google Scholar] [CrossRef]
- Gabriel, C.M.; Hughes, R.A.; Moore, S.E.; Smith, K.J.; Walsh, F.S. Induction of experimental autoimmune neuritis with peripheral myelin protein-22. Brain J. Neurol. 1998, 121 Pt 10, 1895–1902. [Google Scholar] [CrossRef] [Green Version]
- Suzumura, A.; Sobue, G.; Sugimura, K.; Matsuoka, Y.; Sobue, I. Chronic experimental allergic neuritis (EAN) in juvenile guinea pigs: Immunological comparison with acute EAN in adult guinea pigs. Acta Neurol. Scand. 1985, 71, 364–372. [Google Scholar] [CrossRef]
- Harvey, G.K.; Pollard, J.D.; Schindhelm, K.; Antony, J. Chronic experimental allergic neuritis. An electrophysiological and histological study in the rabbit. J. Neurol. Sci. 1987, 81, 215–225. [Google Scholar] [CrossRef]
- Shy, M.E.; Arroyo, E.; Sladky, J.; Menichella, D.; Jiang, H.; Xu, W.; Kamholz, J.; Scherer, S.S. Heterozygous P0 knockout mice develop a peripheral neuropathy that resembles chronic inflammatory demyelinating polyneuropathy (CIDP). J. Neuropathol. Exp. Neurol. 1997, 56, 811–821. [Google Scholar] [CrossRef]
- Jung, S.; Gaupp, S.; Korn, T.; Köllner, G.; Hartung, H.-P.; Toyka, K.V. Biphasic form of experimental autoimmune neuritis in dark Agouti rats and its oral therapy by antigen-specific tolerization. J. Neurosci. Res. 2004, 75, 524–535. [Google Scholar] [CrossRef]
- Salomon, B.; Rhee, L.; Bour-Jordan, H.; Hsin, H.; Montag, A.; Soliven, B.; Arcella, J.; Girvin, A.M.; Miller, S.D.; Bluestone, J.A. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-deficient NOD mice. J. Exp. Med. 2001, 194, 677–684. [Google Scholar] [CrossRef]
- Soliven, B. Autoimmune neuropathies: Insights from animal models. J. Peripher. Nerv. Syst. JPNS 2012, 17 (Suppl. S2), 28–33. [Google Scholar] [CrossRef]
- Ubogu, E.E.; Yosef, N.; Xia, R.H.; Sheikh, K.A. Behavioral, electrophysiological, and histopathological characterization of a severe murine chronic demyelinating polyneuritis model. J. Peripher. Nerv. Syst. JPNS 2012, 17, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Brun, S.; Beaino, W.; Kremer, L.; Taleb, O.; Mensah-Nyagan, A.G.; Lam, C.D.; Greer, J.M.; de Seze, J.; Trifilieff, E. Characterization of a new rat model for chronic inflammatory demyelinating polyneuropathies. J. Neuroimmunol. 2015, 278, 1–10. [Google Scholar] [CrossRef]
- de Sèze, J.; Kremer, L.; Alves do Rego, C.; Taleb, O.; Lam, D.; Beiano, W.; Mensah-Nyagan, G.; Trifilieff, E.; Brun, S. Chronic inflammatory demyelinating polyradiculoneuropathy: A new animal model for new therapeutic targets. Rev. Neurol. 2016, 172, 767–769. [Google Scholar] [CrossRef]
- Brun, S.; Schall, N.; Bonam, S.R.; Bigaut, K.; Mensah-Nyagan, A.-G.; de Sèze, J.; Muller, S. An autophagy-targeting peptide to treat chronic inflammatory demyelinating polyneuropathies. J. Autoimmun. 2018, 92, 114–125. [Google Scholar] [CrossRef]
- Kremer, L.; Taleb, O.; Boehm, N.; Mensah-Nyagan, A.G.; Trifilieff, E.; de Seze, J.; Brun, S. FTY720 controls disease severity and attenuates sciatic nerve damage in chronic experimental autoimmune neuritis. J. Neuroinflamm. 2019, 16, 54. [Google Scholar] [CrossRef]
- Muller, S.; Brun, S.; René, F.; de Sèze, J.; Loeffler, J.-P.; Jeltsch-David, H. Autophagy in neuroinflammatory diseases. Autoimmun. Rev. 2017, 16, 856–874. [Google Scholar] [CrossRef]
- Brun, S.; Schall, N.; Jeltsch-David, H.; Sèze, J.; de Muller, S. Assessing Autophagy in Sciatic Nerves of a Rat Model that Develops Inflammatory Autoimmune Peripheral Neuropathies. Cells 2017, 6, E30. [Google Scholar] [CrossRef] [Green Version]
- Gelinas, D.; Katz, J.; Nisbet, P.; England, J.D. Current practice patterns in CIDP: A cross-sectional survey of neurologists in the United States. J. Neurol. Sci. 2019, 397, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Roessler, H.I.; Knoers, N.V.A.M.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef]
- Gelfand, E.W. Differences between IGIV products: Impact on clinical outcome. Int. Immunopharmacol. 2006, 6, 592–599. [Google Scholar] [CrossRef]
- Bright, R.J.; Wilkinson, J.; Coventry, B.J. Therapeutic options for chronic inflammatory demyelinating polyradiculoneuropathy: A systematic review. BMC Neurol. 2014, 14, 26. [Google Scholar] [CrossRef] [Green Version]
- Casertano, S.; Signoriello, E.; Rossi, F.; Di Pietro, A.; Tuccillo, F.; Bonavita, S.; Lus, G. Ocrelizumab in a case of refractory chronic inflammatory demyelinating polyneuropathy with anti-rituximab antibodies. Eur. J. Neurol. 2020, 27, 2673–2675. [Google Scholar] [CrossRef]
- Li, X.-L.; Dou, Y.-C.; Liu, Y.; Shi, C.-W.; Cao, L.-L.; Zhang, X.-Q.; Zhu, J.; Duan, R.-S. Atorvastatin ameliorates experimental autoimmune neuritis by decreased Th1/Th17 cytokines and up-regulated T regulatory cells. Cell. Immunol. 2011, 271, 455–461. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.-Y.; Fauser, U.; Schluesener, H.J. FTY720 ameliorates experimental autoimmune neuritis by inhibition of lymphocyte and monocyte infiltration into peripheral nerves. Exp. Neurol. 2008, 210, 681–690. [Google Scholar] [CrossRef]
- Vallat, J.-M.; Mathis, S.; Ghorab, K.; Milor, M.-A.; Richard, L.; Magy, L. Natalizumab as a Disease-Modifying Therapy in Chronic Inflammatory Demyelinating Polyneuropathy—A Report of Three Cases. Eur. Neurol. 2015, 73, 294–302. [Google Scholar] [CrossRef]
- Dong, C.; Greathouse, K.M.; Beacham, R.L.; Palladino, S.P.; Helton, E.S.; Ubogu, E.E. Fibronectin connecting segment-1 peptide inhibits pathogenic leukocyte trafficking and inflammatory demyelination in experimental models of chronic inflammatory demyelinating polyradiculoneuropathy. Exp. Neurol. 2017, 292, 35–45. [Google Scholar] [CrossRef]
- Bril, V.; Benatar, M.; Andersen, H.; Vissing, J.; Brock, M.; Greve, B.; Kiessling, P.; Woltering, F.; Griffin, L.; Bergh, P.V.D. Efficacy and Safety of Rozanolixizumab in Moderate to Severe Generalized Myasthenia Gravis: A Phase 2 Randomized Control Trial. Neurology 2021, 96, e853–e865. [Google Scholar] [CrossRef]
- Huijbers, M.G.; Plomp, J.J.; van Es, I.E.; Fillié-Grijpma, Y.E.; Kamar-Al Majidi, S.; Ulrichts, P.; de Haard, H.; Hofman, E.; van der Maarel, S.M.; Verschuuren, J.J. Efgartigimod improves muscle weakness in a mouse model for muscle-specific kinase myasthenia gravis. Exp. Neurol. 2019, 317, 133–143. [Google Scholar] [CrossRef]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J. Clin. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef] [Green Version]
- Halstead, S.K.; Zitman, F.M.P.; Humphreys, P.D.; Greenshields, K.; Verschuuren, J.J.; Jacobs, B.C.; Rother, R.P.; Plomp, J.J.; Willison, H.J. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain J. Neurol. 2008, 131, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Meyer zu Hörste, G.; Mausberg, A.K.; Korth, C.; Stüve, O.; Kieseier, B.C. Quinpramine--a promising compound for treating immune-mediated demyelination of the nervous system. Drug News Perspect. 2010, 23, 287–294. [Google Scholar] [CrossRef]
- Page, N.; Gros, F.; Schall, N.; Décossas, M.; Bagnard, D.; Briand, J.-P.; Muller, S. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann. Rheum. Dis. 2011, 70, 837–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, N.; Gros, F.; Schall, N.; Briand, J.-P.; Muller, S. A therapeutic peptide in lupus alters autophagic processes and stability of MHCII molecules in MRL/lpr B cells. Autophagy 2011, 7, 539–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macri, C.; Wang, F.; Tasset, I.; Schall, N.; Page, N.; Briand, J.-P.; Cuervo, A.M.; Muller, S. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy 2015, 11, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Wang, F.; Schall, N.; Muller, S. Rescue of autophagy and lysosome defects in salivary glands of MRL/lpr mice by a therapeutic phosphopeptide. J. Autoimmun. 2018, 90, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Wang, F.; Schall, N.; Kleinmann, J.-F.; Faludi, M.; Nashi, E.P.; Sibilia, J.; Martin, T.; Schaeffer, E.; Muller, S. Lupus Regulator Peptide P140 Represses B Cell Differentiation by Reducing HLA Class II Molecule Overexpression. Arthritis Rheumatol. 2018, 70, 1077–1088. [Google Scholar] [CrossRef]
- Schall, N.; Page, N.; Macri, C.; Chaloin, O.; Briand, J.-P.; Muller, S. Peptide-based approaches to treat lupus and other autoimmune diseases. J. Autoimmun. 2012, 39, 143–153. [Google Scholar] [CrossRef]
- Zimmer, R.; Scherbarth, H.R.; Rillo, O.L.; Gomez-Reino, J.J.; Muller, S. Lupuzor/P140 peptide in patients with systemic lupus erythematosus: A randomised, double-blind, placebo-controlled phase IIb clinical trial. Ann. Rheum. Dis. 2013, 72, 1830–1835. [Google Scholar] [CrossRef]
- Voynova, E.; Lefebvre, F.; Qadri, A.; Muller, S. Correction of autophagy impairment inhibits pathology in the NOD.H-2h4 mouse model of primary Sjögren’s syndrome. J. Autoimmun. 2020, 108, 102418. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, F.; Schall, N.; Petit-Demoulière, N.; Frossard, N.; Muller, S. An Autophagy Modulator Peptide Prevents Lung Function Decrease and Corrects Established Inflammation in Murine Models of Airway Allergy. Cells 2021, 10, 2468. [Google Scholar] [CrossRef]
- Wang, F.; Tasset, I.; Cuervo, A.M.; Muller, S. In Vivo Remodeling of Altered Autophagy-Lysosomal Pathway by a Phosphopeptide in Lupus. Cells 2020, 9, 2328. [Google Scholar] [CrossRef]
- Gros, F.; Arnold, J.; Page, N.; Décossas, M.; Korganow, A.-S.; Martin, T.; Muller, S. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy 2012, 8, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-Q.; Zhang, J.; Zhou, G. Autophagy and its implication in human oral diseases. Autophagy 2017, 13, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug. Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.Y.; Shin, Y.K.; Park, S.Y.; Park, J.Y.; Rha, S.-H.; Kim, J.K.; Lee, H.J.; Park, H.T. Autophagy is involved in the reduction of myelinating Schwann cell cytoplasm during myelin maturation of the peripheral nerve. PLoS ONE 2015, 10, e0116624. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Chen, X.; Xue, R.; Zhou, Q.; Hu, P.; Ouyang, X.; Dai, T.; Zhu, W.; Tian, S. Autophagy is involved in the pathogenesis of experimental autoimmune neuritis in rats. Neuroreport 2016, 27, 337–344. [Google Scholar] [CrossRef]
- Dalakas, M.C. Potential biomarkers for monitoring therapeutic response in patients with CIDP. J. Peripher. Nerv. Syst. JPNS 2011, 16 (Suppl. S1), 63–67. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Animals | Antigen | Model of Disease | Ref. |
|---|---|---|---|
| Adult guinea pigs | MBP | Acute | [67] |
| Rabbits | Sciatic nerve tissue | Acute | [62] |
| C57BL6 mice | P0(180–199) | Acute | [63,64] |
| C57BL6 mice | P0(106–125) | Acute | [70] |
| SJL mice | P2 protein | Acute | [71] |
| SJL mice | BPNM | Acute | [65] |
| Lewis rats | P2(53–78) | Acute | [72] |
| Lewis rats | P0(180–199) | Acute | [66] |
| Lewis rats | PMP22 | Acute | [73] |
| Juvenile guinea pigs | BPN homogenate | Chronic | [74] |
| Rabbits | BPNM | Chronic | [75] |
| Heterozygous KO (P0+/-) mice | Inherited | Chronic | [76] |
| Dark Agouti rats | BPNM | Chronic | [77] |
| B7-2 KO NOD mice | Spontaneous | Chronic | [78] |
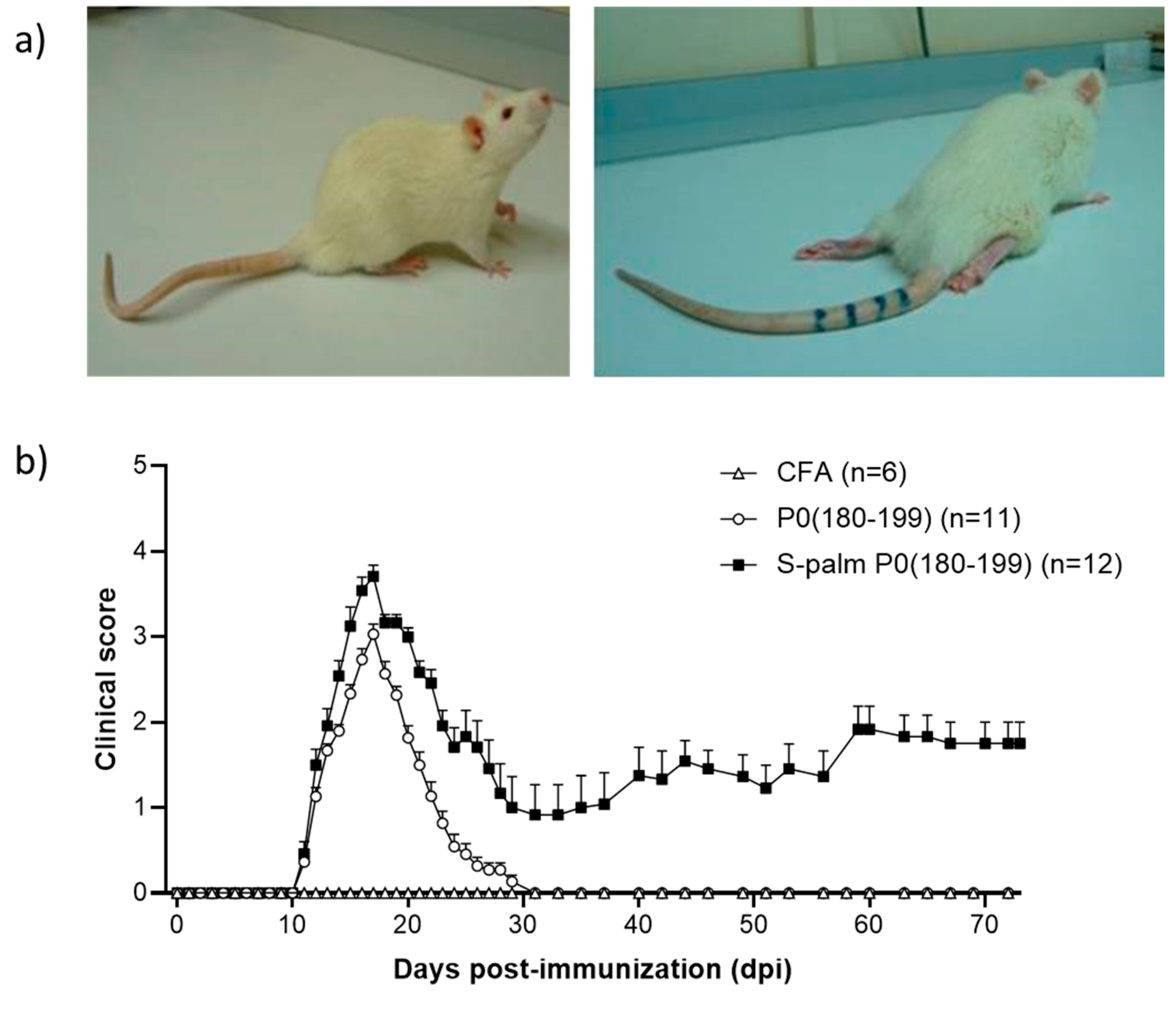
| Lewis rats | S-palm P0(180–199) | Chronic | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brun, S.; de Sèze, J.; Muller, S. CIDP: Current Treatments and Identification of Targets for Future Specific Therapeutic Intervention. Immuno 2022, 2, 118-131. https://doi.org/10.3390/immuno2010009
Brun S, de Sèze J, Muller S. CIDP: Current Treatments and Identification of Targets for Future Specific Therapeutic Intervention. Immuno. 2022; 2(1):118-131. https://doi.org/10.3390/immuno2010009
Chicago/Turabian StyleBrun, Susana, Jérôme de Sèze, and Sylviane Muller. 2022. "CIDP: Current Treatments and Identification of Targets for Future Specific Therapeutic Intervention" Immuno 2, no. 1: 118-131. https://doi.org/10.3390/immuno2010009
APA StyleBrun, S., de Sèze, J., & Muller, S. (2022). CIDP: Current Treatments and Identification of Targets for Future Specific Therapeutic Intervention. Immuno, 2(1), 118-131. https://doi.org/10.3390/immuno2010009