

Computational Screening of Approved Drugs for Inhibition of the Antibiotic Resistance Gene mecA in Methicillin-Resistant Staphylococcus aureus (MRSA) Strains


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mining and Downloading of MecA and Blasting against the DrugBank Database
2.2. Modeling and Docking Experiment
2.3. Search for Structural Similarities to Known Antibiotics and High-Affinity Docking
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Algammal, A.M.; Hetta, H.F.; Elkelish, A.; Alkhalifah, D.H.H.; Hozzein, W.N.; Batiha, G.E.S.; Nahhas, N.E.; Mabrok, M.A. Methicillin-Resistant Staphylococcus aureus (MRSA): One Health Perspective Approach to the Bacterium Epidemiology, Virulence Factors, Antibiotic-Resistance, and Zoonotic Impact. Infect. Drug Resist. 2020, 13, 3255–3265. [Google Scholar] [CrossRef]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G., Jr. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Panesso, D.; Planet, P.J.; Diaz, L.; Hugonnet, J.E.; Tran, T.T.; Narechania, A.; Munita, J.M.; Rincon, S.; Carvajal, L.P.; Reyes, J.; et al. Methicillin-susceptible, vancomycin-resistant Staphylococcus aureus, Brazil. Emerg. Infect. Dis. 2015, 21, 1844. [Google Scholar] [CrossRef]
- Rehm, S.J.; Tice, A. Staphylococcus aureus: Methicillin-susceptible S. aureus to methicillin-resistant S. aureus and vancomycin-resistant S. aureus. Clin. Infect. Dis. 2010, 51 (Suppl. S2), S176–S182. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; De Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Prim. 2018, 4, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention (CDC). Deadly Staph Infections Still Threaten the US; Retrieved November; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2019; Volume 13, p. 2021.
- DeLeo, F.R.; Chambers, H.F. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin. Investig. 2009, 119, 2464–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, T.; Geoghegan, J. Chapter 37—Staphylococcus aureus. In Molecular Medical Microbiology, 2nd ed.; Tang, Y.-W., Sussman, M., Liu, D., Poxton, I., Schwartzman, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 655–674. [Google Scholar]
- Liu, W.T.; Chen, E.Z.; Yang, L.; Peng, C.; Wang, Q.; Xu, Z.; Chen, D.Q. Emerging resistance mechanisms for 4 types of common anti-MRSA antibiotics in Staphylococcus aureus: A comprehensive review. Microb. Pathog. 2021, 156, 104915. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Chen, E.Z.; Yang, L.; Peng, C.; Wang, Q.; Xu, Z.; Chen, D.Q. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef]
- Peyclit, L.; Baron, S.A.; Rolain, J.-M. Drug repurposing to fight colistin and carbapenem-resistant bacteria. Front. Cell. Infect. Microbiol. 2019, 9, 193. [Google Scholar] [CrossRef]
- Prasannanjaneyulu, V.; Nene, S.; Jain, H.; Nooreen, R.; Otavi, S.; Chitlangya, P.; Srivastava, S. Old drugs, new tricks: Emerging role of drug repurposing in the management of atopic dermatitis. Cytokine Growth Factor Rev. 2022, 65, 12–26. [Google Scholar] [CrossRef]
- Savoia, D. New antimicrobial approaches: Reuse of old drugs. Curr. Drug Targets 2016, 17, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, R.; Colmer, S.; Tynan, H.; Demain, A.L.; Gullo, V.P. Antimicrobials, drug discovery, and genome mining. Appl. Microbiol. Biotechnol. 2013, 97, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Weese, J.S. Methicillin-resistant Staphylococcus aureus in animals. ILAR J. 2010, 51, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Graveland, H.; Wagenaar, J.A.; Bergs, K.; Heesterbeek, H.; Heederik, D. Persistence of livestock associated MRSA CC398 in humans is dependent on intensity of animal contact. PLoS ONE 2011, 6, e16830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorado-García, A.; Bos, M.E.; Graveland, H.; Van Cleef, B.A.; Verstappen, K.M.; Kluytmans, J.A.; Wagenaar, J.A.; Heederik, D.J. Risk factors for persistence of livestock-associated MRSA and environmental exposure in veal calf farmers and their family members: An observational longitudinal study. BMJ Open 2013, 3, e003272. [Google Scholar] [CrossRef] [Green Version]
- Choo, E.J.; Chambers, H.F. Treatment of methicillin-resistant Staphylococcus aureus bacteremia. Infect. Chemother. 2016, 48, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Lakhundi, S.; Zhang, K. Methicillin-resistant Staphylococcus aureus: Molecular characterization, evolution, and epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18. [Google Scholar] [CrossRef] [Green Version]
- Baëtz, B.; Boudrioua, A.; Hartke, A.; Giraud, C. Alternatives to fight vancomycin-resistant staphylococci and enterococci. Antibiotics 2021, 10, 1116. [Google Scholar] [CrossRef]
- Otarigho, B.; Falade, M.O. Analysis of antibiotics resistant genes in different strains of Staphylococcus aureus. Bioinformation 2018, 14, 113. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, H. Staphylococcus: Skin Infections; Osteomyelitis; Bloodstream Infection; Food Poisoning; Foreign Body Infections; MRSA, in Medical Microbiology; Elsevier: Amsterdam, The Netherlands, 2012; pp. 176–182. [Google Scholar]
- Tsubakishita, S.; Kuwahara-Arai, K.; Sasaki, T.; Hiramatsu, K. Origin and molecular evolution of the determinant of methicillin resistance in staphylococci. Antimicrob. Agents Chemother. 2010, 54, 4352–4359. [Google Scholar] [CrossRef] [Green Version]
- Gootz, T.D. Discovery and development of new antimicrobial agents. Clin. Microbiol. Rev. 1990, 3, 13–31. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Lui, H.K.; Gao, W.; Cheung, K.C.; Jin, W.B.; Sun, N.; Kan, J.W.; Wong, I.L.; Chiou, J.; Lin, D.; Chan, E.W.; et al. Boosting the efficacy of anti-MRSA β-lactam antibiotics via an easily accessible, non-cytotoxic and orally bioavailable FtsZ inhibitor. Eur. J. Med. Chem. 2019, 163, 95–115. [Google Scholar] [CrossRef] [PubMed]
- An, J.X.; Ma, Y.; Zhao, W.B.; Hu, Y.M.; Wang, Y.R.; Zhang, Z.J.; Luo, X.F.; Zhang, B.Q.; Ding, Y.Y.; Liu, Y.Q. Drug repurposing strategy II: From approved drugs to agri-fungicide leads. J. Antibiot. 2023, 76, 131–182. [Google Scholar] [CrossRef]
- Trezza, A.; Iovinelli, D.; Santucci, A.; Prischi, F.; Spiga, O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020, 10, 13866. [Google Scholar] [CrossRef] [PubMed]
- Gieringer, J.H.; Wenz, A.F.; Just, H.M.; Daschner, F.D. Effect of 5-fluorouracil, mitoxantrone, methotrexate, and vincristine on the antibacterial activity of ceftriaxone, ceftazidime, cefotiam, piperacillin, and netilmicin. Chemotherapy 1986, 32, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Minandri, F.; Bonchi, C.; Frangipani, E.; Imperi, F.; Visca, P. Promises and failures of gallium as an antibacterial agent. Future Microbiol. 2014, 9, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Carroni, M.; Franke, K.B.; Maurer, M.; Jäger, J.; Hantke, I.; Gloge, F.; Linder, D.; Gremer, S.; Turgay, K.; Bukau, B.; et al. Regulatory coiled-coil domains promote head-to-head assemblies of AAA+ chaperones essential for tunable activity control. Elife 2017, 6, e30120. [Google Scholar] [CrossRef]
- Lim, D.; Strynadka, N.C. Structural basis for the β lactam resistance of PBP2a from methicillin-resistant Staphylococcus aureus. Nat. Struct. Biol. 2002, 9, 870–876. [Google Scholar] [CrossRef]
- Safo, M.K.; Ko, T.P.; Musayev, F.N.; Zhao, Q.; Wang, A.J.; Archer, G.L. Structure of the MecI repressor from Staphylococcus aureus in complex with the cognate DNA operator of mec. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 320–324. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Gan, J.; Chen, S.; Xiao, Z.X.; Cao, Y. CB-Dock2: Improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022, 50, W159–W164. [Google Scholar] [CrossRef]
- Köck, R.; Becker, K.; Cookson, B.; van Gemert-Pijnen, J.E.; Harbarth, S.; Kluytmans, J.A.J.W.; Mielke, M.; Peters, G.; Skov, R.L.; Struelens, M.J.; et al. Methicillin-resistant Staphylococcus aureus (MRSA): Burden of disease and control challenges in Europe. Eurosurveillance 2010, 15, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefani, S.; Chung, D.R.; Lindsay, J.A.; Friedrich, A.W.; Kearns, A.M.; Westh, H.; MacKenzie, F.M. Meticillin-resistant Staphylococcus aureus (MRSA): Global epidemiology and harmonisation of typing methods. Int. J. Antimicrob. Agents 2012, 39, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Ubukata, K.; Nonoguchi, R.; Matsuhashi, M.; Konno, M. Expression and inducibility in Staphylococcus aureus of the mecA gene, which encodes a methicillin-resistant S. aureus-specific penicillin-binding protein. J. Bacteriol. 1989, 171, 2882–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boswihi, S.S.; Udo, E.E. Methicillin-resistant Staphylococcus aureus: An update on the epidemiology, treatment options and infection control. Curr. Med. Res. Pract. 2018, 8, 18–24. [Google Scholar] [CrossRef]
- Willis, J.A.; Ubukata, K.; Nonoguchi, R.; Matsuhashi, M.; Konno, M. Breaking down antibiotic resistance in methicillin-resistant Staphylococcus aureus: Combining antimicrobial photodynamic and antibiotic treatments. Proc. Natl. Acad. Sci. USA 2022, 119, e2208378119. [Google Scholar] [CrossRef]
- De, P.; Kumar, V.; Kar, S.; Roy, K.; Leszczynski, J. Repurposing FDA approved drugs as possible anti-SARS-CoV-2 medications using ligand-based computational approaches: Sum of ranking difference-based model selection. Struct. Chem. 2022, 33, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, C.; Murugan, N.A.; Priyakumar, U.D. Structure-based drug repurposing: Traditional and advanced AI/ML-aided methods. Drug Discov. Today 2022, 27, 1847–1861. [Google Scholar] [CrossRef] [PubMed]
- Miró-Canturri, A.; Ayerbe-Algaba, R.; Smani, Y. Drug repurposing for the treatment of bacterial and fungal infections. Front. Microbiol. 2019, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, M.A.W.; Dokla, E.M.; Serya, R.A.; Abouzid, K.A. Penicillin binding protein 2a: An overview and a medicinal chemistry perspective. Eur. J. Med. Chem. 2020, 199, 112312. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.N.; Barry, A.L. Cefoperazone: A review of its antimicrobial spectrum, β-lactamase stability, enzyme inhibition, and other in vitro characteristics. Rev. Infect. Dis. 1983, 5 (Suppl. S1), S108–S126. [Google Scholar] [CrossRef]
- Matsubara, N.; Minami, S.; Matsuhashi, M.; Takaoka, M.; Mitsuhashi, S. Affinity of cefoperazone for penicillin-binding proteins. Antimicrob. Agents Chemother. 1980, 18, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Kristof, R.A.; Clusmann, H.; Koehler, W.; Fink, K.B.; Schramm, J. Treatment of accidental high dose intraventricular mezlocillin application by cerebrospinal fluid exchange. J. Neurol. Neurosurg. Psychiatry 1998, 64, 379–381. [Google Scholar] [CrossRef]
- Wang, H.; Yu, Y.; Xie, X.; Wang, C.; Zhang, Y.; Yuan, Y.; Zhang, X.; Liu, J.; Wang, P.; Chen, M. In-vitro antibacterial activities of cefpiramide and other broad-spectrum antibiotics against 440 clinical isolates in China. J. Infect. Chemother. 2000, 6, 81–85. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Chung, P.; Adam, H.; Zelenitsky, S.; Denisuik, A.; Schweizer, F.; Lagacé-Wiens, P.R.; Rubinstein, E.; Gin, A.S.; Walkty, A.; et al. Ceftolozane/tazobactam: A novel cephalosporin/β-lactamase inhibitor combination with activity against multidrug-resistant gram-negative bacilli. Drugs 2014, 74, 31–51. [Google Scholar] [CrossRef]
- Miller, B.; Hershberger, E.; Benziger, D.; Trinh, M.; Friedland, I. Pharmacokinetics and safety of intravenous ceftolozane-tazobactam in healthy adult subjects following single and multiple ascending doses. Antimicrob. Agents Chemother. 2012, 56, 3086–3091. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.K.; Mercer, D.; Itani, K.M.; Nicolau, D.P.; Kuti, J.L.; Mansfield, D.; Dana, A. Randomized, open-label, comparative study of piperacillin-tazobactam administered by continuous infusion versus intermittent infusion for treatment of hospitalized patients with complicated intra-abdominal infection. Antimicrob. Agents Chemother. 2006, 50, 3556–3561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Roberts, J.A.; Paterson, D.L.; Lipman, J. Pharmacokinetic evaluation of piperacillin-tazobactam. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Nix, D.E.; Majumdar, A.K.; DiNubile, M.J. Pharmacokinetics and pharmacodynamics of ertapenem: An overview for clinicians. J. Antimicrob. Chemother. 2004, 53 (Suppl. S2), ii23–ii28. [Google Scholar] [CrossRef]
- de Vries, H.J.; de Laat, M.; Jongen, V.W.; Heijman, T.; Wind, C.M.; Boyd, A.; de Korne-Elenbaas, J.; van Dam, A.P.; van der Loeff, M.F.S.; Bruisten, S.; et al. Efficacy of ertapenem, gentamicin, fosfomycin, and ceftriaxone for the treatment of anogenital gonorrhoea (NABOGO): A randomised, non-inferiority trial. Lancet Infect. Dis. 2022, 22, 706–717. [Google Scholar] [CrossRef]
- French, G. Bactericidal agents in the treatment of MRSA infections—The potential role of daptomycin. J. Antimicrob. Chemother. 2006, 58, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Kampf, G.; Jarosch, R.; Rüden, H. Limited effectiveness of chlorhexidine based hand disinfectants against methicillin-resistant Staphylococcus aureus (MRSA). J. Hosp. Infect. 1998, 38, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Biedenbach, D.J.; Arhin, F.F.; Moeck, G.; Lynch, T.F.; Sahm, D.F. In vitro activity of oritavancin and comparator agents against staphylococci, streptococci and enterococci from clinical infections in Europe and North America, 2011–2014. Int. J. Antimicrob. Agents 2015, 46, 674–681. [Google Scholar] [CrossRef]
- Kim, S.J.; Cegelski, L.; Stueber, D.; Singh, M.; Dietrich, E.; Tanaka, K.S.; Parr, T.R., Jr.; Far, A.R.; Schaefer, J. Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis in Staphylococcus aureus. J. Mol. Biol. 2008, 377, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Zhanel, G.G.; Schweizer, F.; Karlowsky, J.A. Oritavancin: Mechanism of action. Clin. Infect. Dis. 2012, 54 (Suppl. S3), S214–S219. [Google Scholar] [CrossRef] [Green Version]
- Balwani, M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: Pathophysiology, genetics, clinical manifestations, and management. Mol. Genet. Metab. 2019, 128, 298–303. [Google Scholar] [CrossRef]
- Minder, E.I.; Barman-Aksoezen, J.; Schneider-Yin, X. Pharmacokinetics and pharmacodynamics of afamelanotide and its clinical use in treating dermatologic disorders. Clin. Pharmacokinet. 2017, 56, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Garnock-Jones, K.P. Afamelanotide: A review in erythropoietic protoporphyria. Am. J. Clin. Dermatol. 2016, 17, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Langendonk, J.G.; Balwani, M.; Anderson, K.E.; Bonkovsky, H.L.; Anstey, A.V.; Bissell, D.M.; Bloomer, J.; Edwards, C.; Neumann, N.J.; Parker, C.; et al. Afamelanotide for erythropoietic protoporphyria. N. Engl. J. Med. 2015, 373, 48–59. [Google Scholar] [CrossRef] [PubMed]







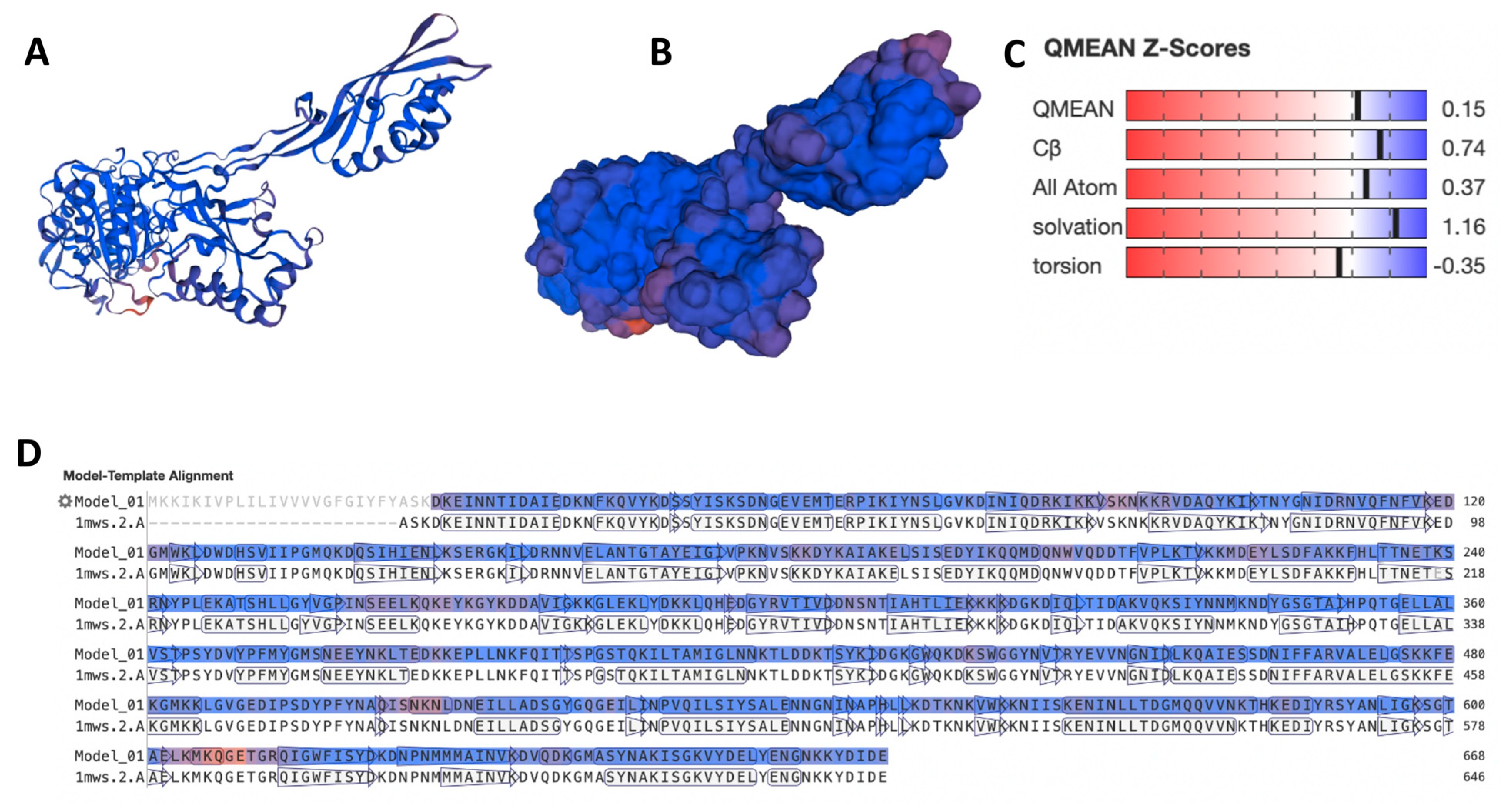{kind=link}
{kind=link}
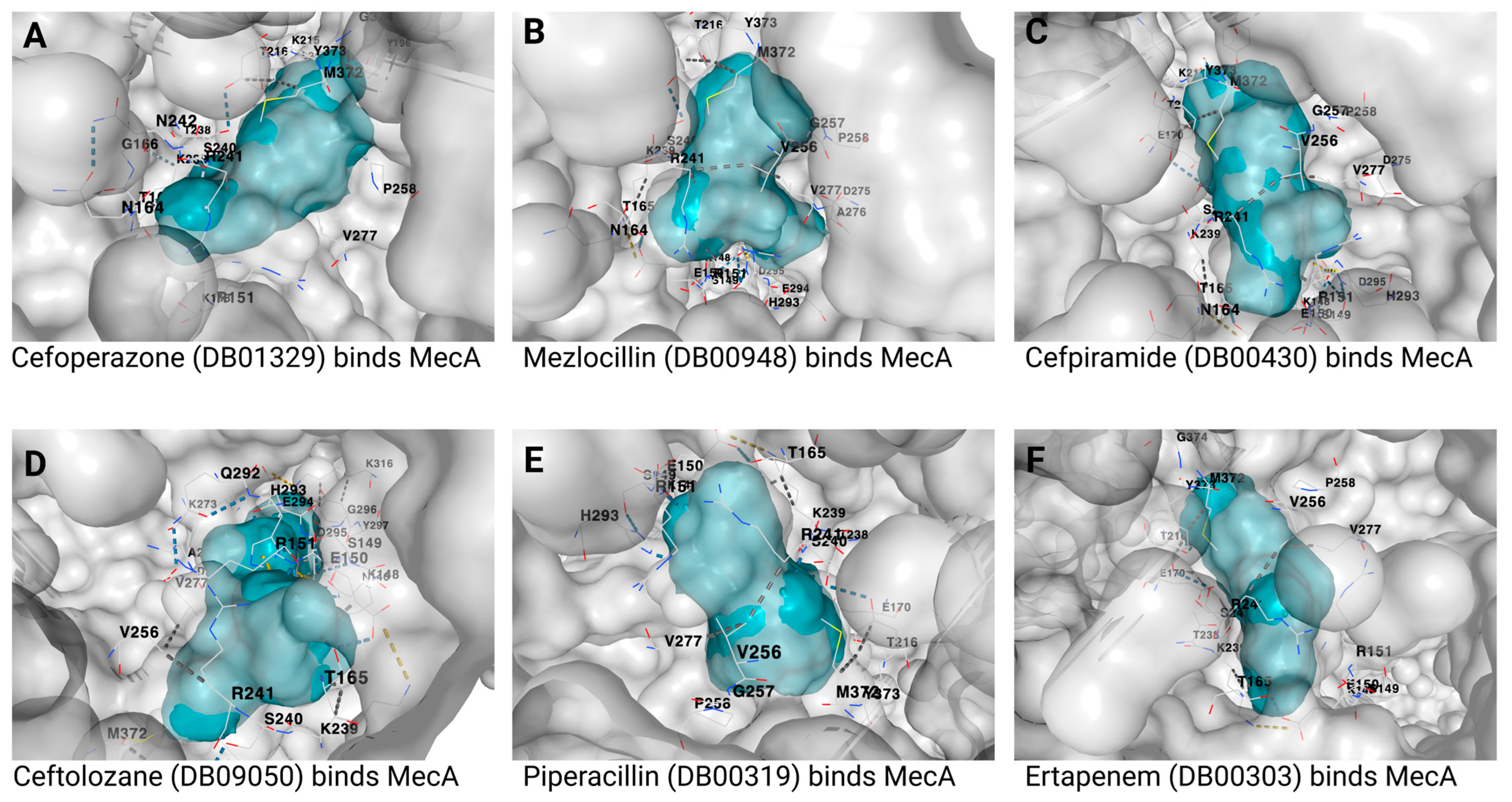{kind=link}
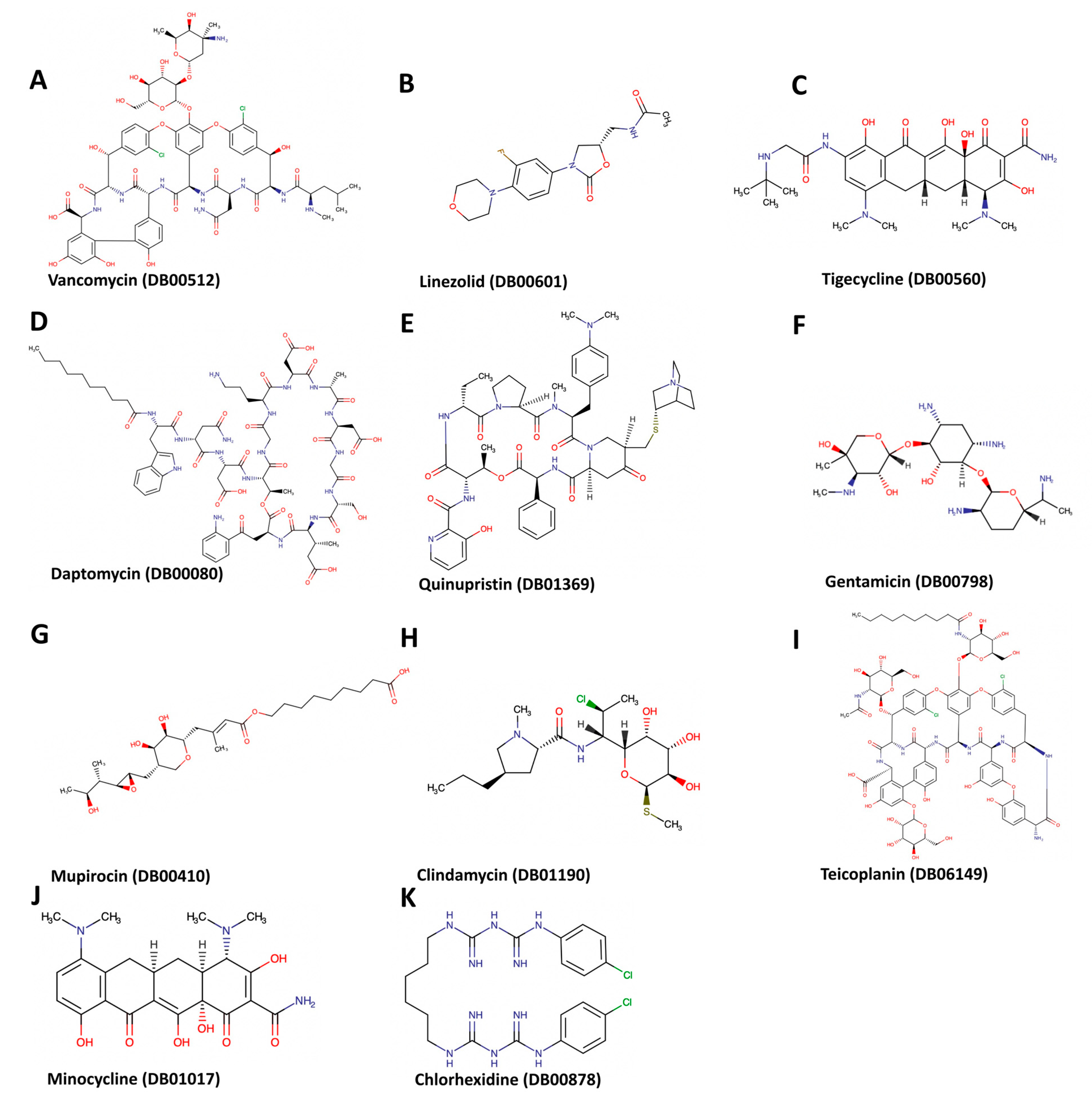{kind=link}
{kind=link}
{kind=link}
| Target | Protein | E Value | Bit Score | Query Length | Alignment Length | No. of Binding Drugs to Each Target |
|---|---|---|---|---|---|---|
| 1 | MecA | 0 | 1273.46 | 668 | 645 | 1 |
| 2 | MecA PBP2′ (penicillin binding protein 2′) | 0 | 1216.83 | 668 | 649 | 2 |
| 3 | Penicillin binding protein 2a | 0 | 979.934 | 668 | 486 | 4 |
| 4 | Penicillin-binding protein 3 | 1.39 × 10−132 | 404.06 | 668 | 532 | 4 |
| 5 | Penicillin-binding protein 3 | 2.63 × 10−121 | 375.17 | 668 | 609 | 1 |
| 6 | Penicillin-binding protein 2 | 6.14 × 10−62 | 199.904 | 668 | 95 | 1 |
| 7 | Penicillin-binding protein 2 | 9.59 × 10−43 | 162.54 | 668 | 553 | 2 |
| 8 | Penicillin-binding protein 2 | 1.19 × 10−38 | 150.599 | 668 | 515 | 17 |
| 9 | Penicillin-binding protein 2 | 7.65 × 10−35 | 139.043 | 668 | 564 | 2 |
| 10 | Penicillin-binding protein 2 | 7.69 × 10−31 | 126.716 | 668 | 530 | 2 |
| 11 | Peptidoglycan synthase FtsI | 1.88 × 10−20 | 94.3597 | 668 | 556 | 18 |
| 12 | Peptidoglycan synthase FtsI | 2.91 × 10−19 | 90.5077 | 668 | 551 | 1 |
| 13 | PBP3 | 1.72 × 10−17 | 85.1149 | 668 | 551 | 1 |
| 14 | Penicillin-binding protein 2 | 3.71 × 10−16 | 80.4925 | 668 | 457 | 1 |
| 15 | Cell division protein | 1.28 × 10−15 | 78.9518 | 668 | 515 | 4 |
| 16 | Penicillin-binding protein 2x | 3.20 × 10−15 | 77.7962 | 668 | 522 | 2 |
| 17 | Penicillin-binding protein 2X | 5.65 × 10−15 | 77.0258 | 668 | 522 | 2 |
| 18 | Penicillin-binding protein 2B | 6.89 × 10−12 | 67.0106 | 668 | 462 | 2 |
| 19 | Penicillin-binding protein 2B | 6.89 × 10−12 | 67.0106 | 668 | 462 | 17 |
| S/N | Generic Name | DrugBank Accession Number | Vina Score | Cavity Volume (Å3) | Center (x, y, z) | Docking Size (x, y, z) |
|---|---|---|---|---|---|---|
| 1 | Cefoperazone | DB01329 | −9.3 | 886 | −3, 43, 37 | 27, 27, 27 |
| 2 | Mezlocillin | DB00948 | −8.8 | 886 | −3, 43, 37 | 23, 23, 23 |
| 3 | Cefpiramide | DB00430 | −8.8 | 886 | −3, 43, 37 | 26, 26, 26 |
| 4 | Ceftolozane | DB09050 | −8.7 | 886 | −3, 43, 37 | 27, 27, 27 |
| 5 | Piperacillin | DB00319 | −8.6 | 886 | −3, 43, 37 | 24, 24, 24 |
| 6 | Ertapenem | DB00303 | −8.5 | 886 | −3, 43, 37 | 23, 23, 23 |
| S/N | Generic Name | DrugBank Accession Number | Vina Score | Cavity Volume (Å3) | Center (x, y, z) | Docking Size (x, y, z) |
|---|---|---|---|---|---|---|
| 1 | Daptomycin | DB00080 | −9.8 | 886 | −3, 43, 37 | 34, 34, 34 |
| 2 | Linezolid | DB00601 | −7.2 | 886 | −3, 43, 37 | 24, 24, 24 |
| 3 | Tigecycline | DB00560 | −8.4 | 886 | −3, 43, 37 | 26, 26, 26 |
| 4 | Quinupristin/Dalfopristin | DB01369 | −8.5 | 863 | −19, 38, 32 | 28, 28, 28 |
| 5 | Vancomycin | DB00512 | −11.6 | 886 | −3, 43, 37 | 32, 32, 32 |
| 6 | Gentamicin | DB00798 | −7.6 | 886 | −3, 43, 37 | 24, 24, 24 |
| 7 | Mupirocin | DB00410 | −7.5 | 886 | −3, 43, 37 | 32, 32, 32 |
| 8 | Minocycline | DB01017 | −7 | 996 | −33, 53, 59 | 22, 22, 22 |
| 9 | Clindamycin | DB01190 | −6.5 | 886 | −3, 43, 37 | 22, 22, 22 |
| 10 | Chlorhexidine | DB00878 | −9.3 | 886 | −3, 43, 37 | 22, 22, 22 |
| 11 | Oritavancin | DB04911 | −10.3 | 886 | −3, 43, 37 | 34, 34, 34 |
| 12 | Telavancin | DB06402 | −8.4 | 863 | −19, 38, 32 | 34, 34, 34 |
| 13 | Afamelanotide | DB04931 | −9.5 | 886 | −3, 43, 37 | 38, 38, 38 |
| 14 | Proguanil | DB01131 | −6.7 | 996 | −33, 53, 59 | 22, 22, 22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otarigho, B.; Falade, M.O. Computational Screening of Approved Drugs for Inhibition of the Antibiotic Resistance Gene mecA in Methicillin-Resistant Staphylococcus aureus (MRSA) Strains. BioTech 2023, 12, 25. https://doi.org/10.3390/biotech12020025
Otarigho B, Falade MO. Computational Screening of Approved Drugs for Inhibition of the Antibiotic Resistance Gene mecA in Methicillin-Resistant Staphylococcus aureus (MRSA) Strains. BioTech. 2023; 12(2):25. https://doi.org/10.3390/biotech12020025
Chicago/Turabian StyleOtarigho, Benson, and Mofolusho O. Falade. 2023. "Computational Screening of Approved Drugs for Inhibition of the Antibiotic Resistance Gene mecA in Methicillin-Resistant Staphylococcus aureus (MRSA) Strains" BioTech 12, no. 2: 25. https://doi.org/10.3390/biotech12020025
APA StyleOtarigho, B., & Falade, M. O. (2023). Computational Screening of Approved Drugs for Inhibition of the Antibiotic Resistance Gene mecA in Methicillin-Resistant Staphylococcus aureus (MRSA) Strains. BioTech, 12(2), 25. https://doi.org/10.3390/biotech12020025