
The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
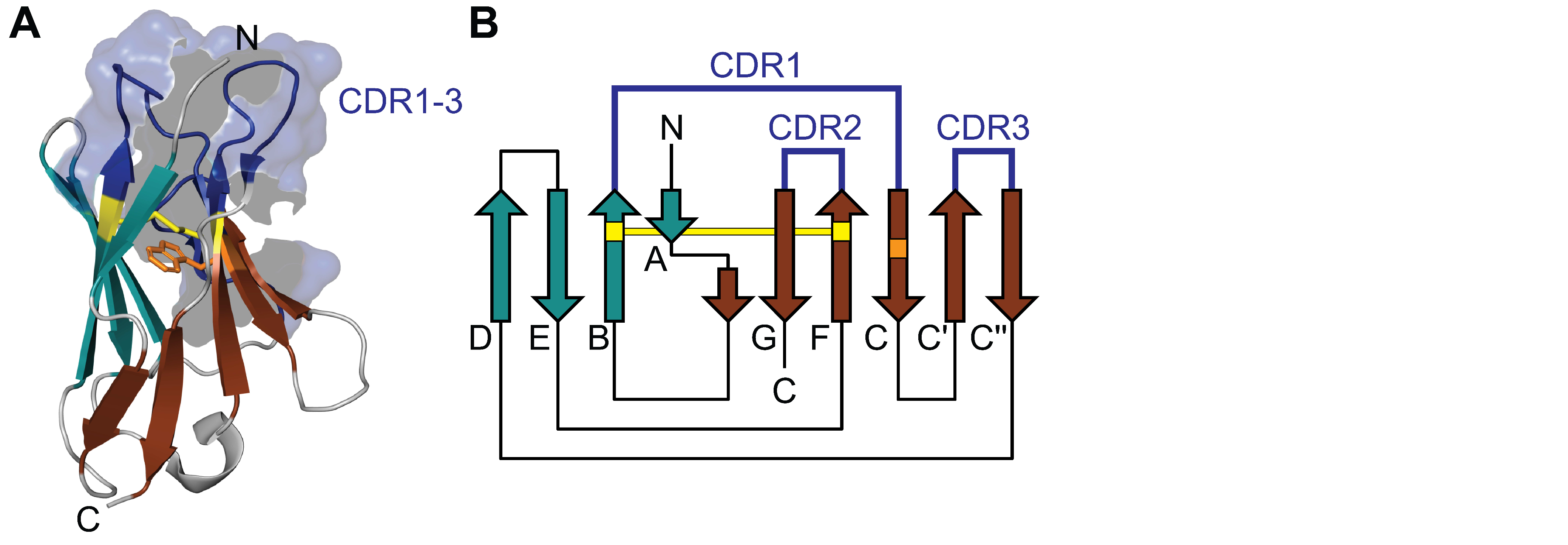
2. Structure and Variability of Light Chains
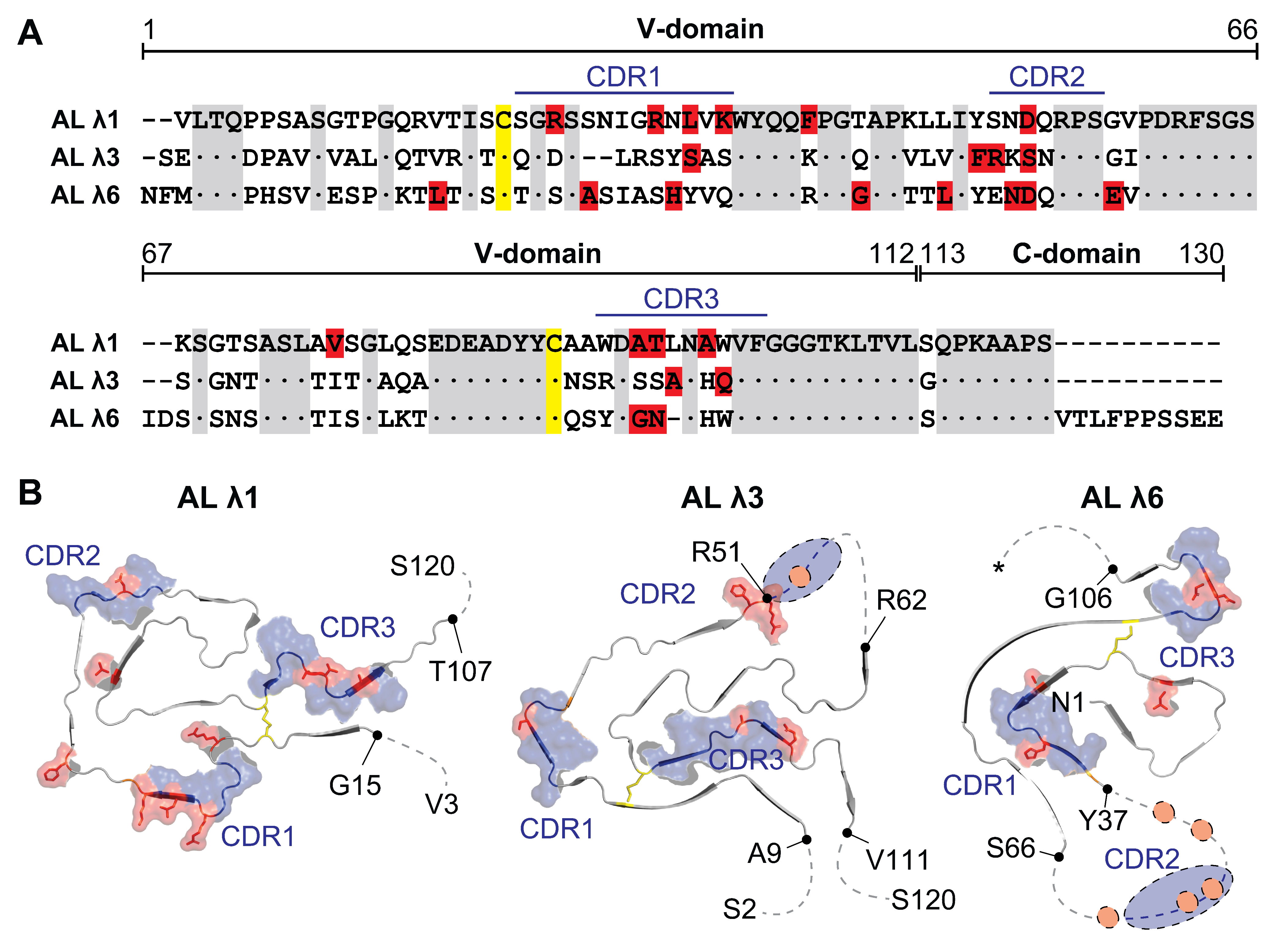
3. Properties of Amyloidogenic Light Chains
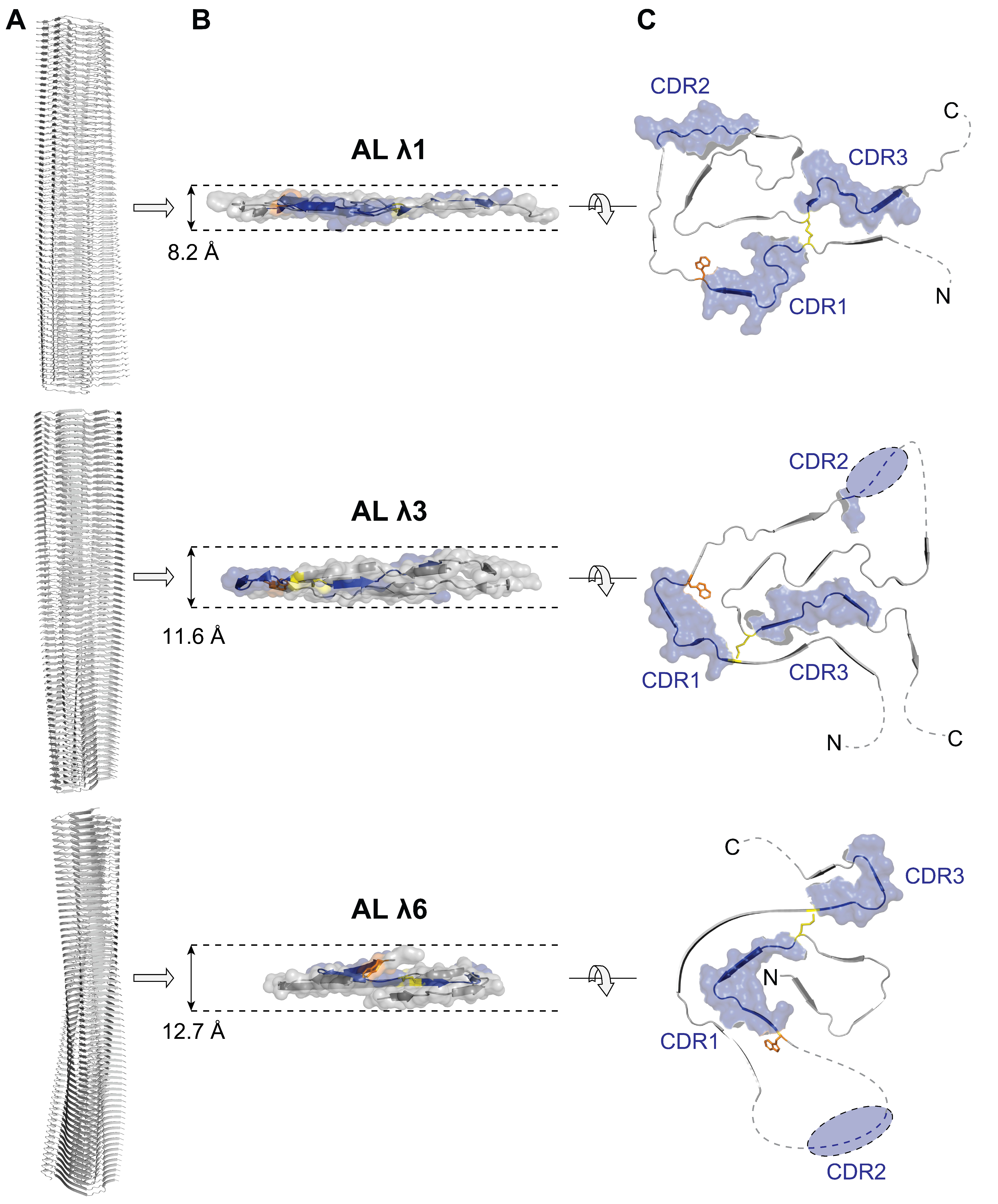
4. Structure of AL Fibrils
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Merlini, G.; Dispenzieri, A.; Sanchorawala, V.; Schönland, S.O.; Palladini, G.; Hawkins, P.N.; Gertz, M.A. Systemic immunoglobulin light chain amyloidosis. Nat. Rev. Dis. Primers 2018, 4, 38. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Bork, P.; Holm, L.; Sander, C. The immunoglobulin fold: Structural classification, sequence patterns and common core. J. Mol. Biol. 1994, 242, 309–320. [Google Scholar] [CrossRef]
- Lefranc, M.P.; Lefranc, G. Immunoglobulins or Antibodies: IMGT® Bridging Genes, Structures and Functions. Biomedicines 2020, 8, 319. [Google Scholar] [CrossRef]
- Bellotti, V.; Mangione, P.; Merlini, G. Review: Immunoglobulin light chain amyloidosis—The archetype of structural and pathogenic variability. J. Struct. Biol. 2000, 130, 280–289. [Google Scholar] [CrossRef]
- Collins, A.M.; Watson, C.T. Immunoglobulin Light Chain Gene Rearrangements, Receptor Editing and the Development of a Self-Tolerant Antibody Repertoire. Front. Immunol. 2018, 9, 2249. [Google Scholar] [CrossRef] [Green Version]
- Odegard, V.H.; Schatz, D.G. Targeting of somatic hypermutation. Nat. Rev. Immunol. 2006, 6, 573–583. [Google Scholar] [CrossRef]
- Herrera, G.A.; Teng, J.; Turbat-Herrera, E.A.; Zeng, C.; del Pozo-Yauner, L. Understanding Mesangial Pathobiology in AL-Amyloidosis and Monoclonal Ig Light Chain Deposition Disease. Kidney Int. Rep. 2020, 5, 1870–1893. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.S.; Geyer, S.M.; Price-Troska, T.L.; Allmer, C.; Kyle, R.A.; Gertz, M.A.; Fonseca, R. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL). Blood 2003, 101, 3801–3808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huang, X.; Li, J. Light chain amyloidosis: Where are the light chains from and how they play their pathogenic role? Blood Rev. 2017, 31, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Townsend, C.L.; Laffy, J.M.; Wu, Y.B.; Silva O’Hare, J.; Martin, V.; Kipling, D.; Fraternali, F.; Dunn-Walters, D.K. Significant Differences in Physicochemical Properties of Human Immunoglobulin Kappa and Lambda CDR3 Regions. Front. Immunol. 2016, 7, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Kant, R.; Bauer, J.; Karow-Zwick, A.R.; Kube, S.; Garidel, P.; Blech, M.; Rousseau, F.; Schymkowitz, J. Adaption of human antibody λ and κ light chain architectures to CDR repertoires. Protein Eng. Des. Sel. 2019, 32, 109–127. [Google Scholar] [CrossRef] [Green Version]
- Comenzo, R.L.; Zhang, Y.; Martinez, C.; Osman, K.; Herrera, G.A. The tropism of organ involvement in primary systemic amyloidosis: Contributions of Ig V(L) germ line gene use and clonal plasma cell burden. Blood 2001, 98, 714–720. [Google Scholar] [CrossRef] [Green Version]
- Perfetti, V.; Casarini, S.; Palladini, G.; Vignarelli, M.C.; Klersy, C.; Diegoli, M.; Ascari, E.; Merlini, G. Analysis of V(lambda)-J(lambda) expression in plasma cells from primary (AL) amyloidosis and normal bone marrow identifies 3r (lambdaIII) as a new amyloid-associated germline gene segment. Blood 2002, 100, 948–953. [Google Scholar] [CrossRef] [PubMed]
- del Pozo-Yauner, L.; Becerril, B.; Ochoa-Leyva, A.; Rodríguez-Ambriz, S.L.; Pérez Carrión, J.I.; Zavala-Padilla, G.; Sánchez-López, R.; Fernández Velasco, D.A. The Structural Determinants of the Immunoglobulin Light Chain Amyloid Aggregation. In Physical Biology of Proteins and Peptides; Olivares-Quiroz, L., Guzmán-López, O., Jardón-Valadez, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 1–28. [Google Scholar]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. 2018, 54, 10664–10674. [Google Scholar] [CrossRef] [PubMed]
- Oberti, L.; Rognoni, P.; Barbiroli, A.; Lavatelli, F.; Russo, R.; Maritan, M.; Palladini, G.; Bolognesi, M.; Merlini, G.; Ricagno, S. Concurrent structural and biophysical traits link with immunoglobulin light chains amyloid propensity. Sci. Rep. 2017, 7, 16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, F.J. Four structural risk factors identify most fibril-forming kappa light chains. Amyloid 2000, 7, 200–211. [Google Scholar] [CrossRef]
- Poshusta, T.L.; Sikkink, L.A.; Leung, N.; Clark, R.J.; Dispenzieri, A.; Ramirez-Alvarado, M. Mutations in specific structural regions of immunoglobulin light chains are associated with free light chain levels in patients with AL amyloidosis. PLoS ONE 2009, 4, e5169. [Google Scholar] [CrossRef] [PubMed]
- Kazman, P.; Vielberg, M.T.; Pulido Cendales, M.D.; Hunziger, L.; Weber, B.; Hegenbart, U.; Zacharias, M.; Köhler, R.; Schönland, S.; Groll, M.; et al. Fatal amyloid formation in a patient’s antibody light chain is caused by a single point mutation. Elife 2020, 9, e52300. [Google Scholar] [CrossRef]
- Rottenaicher, G.J.; Weber, B.; Rührnößl, F.; Kazman, P.; Absmeier, R.M.; Hitzenberger, M.; Zacharias, M.; Buchner, J. Molecular mechanism of amyloidogenic mutations in hypervariable regions of antibody light chains. J. Biol. Chem. 2021, 296, 100334. [Google Scholar] [CrossRef] [PubMed]
- Hurle, M.R.; Helms, L.R.; Li, L.; Chan, W.; Wetzel, R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc. Natl. Acad. Sci. USA 1994, 91, 5446–5450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wall, J.; Schell, M.; Murphy, C.; Hrncic, R.; Stevens, F.J.; Solomon, A. Thermodynamic instability of human lambda 6 light chains: Correlation with fibrillogenicity. Biochemistry 1999, 38, 14101–14108. [Google Scholar] [CrossRef]
- Baden, E.M.; Randles, E.G.; Aboagye, A.K.; Thompson, J.R.; Ramirez-Alvarado, M. Structural insights into the role of mutations in amyloidogenesis. J. Biol. Chem. 2008, 283, 30950–30956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baden, E.M.; Owen, B.A.; Peterson, F.C.; Volkman, B.F.; Ramirez-Alvarado, M.; Thompson, J.R. Altered dimer interface decreases stability in an amyloidogenic protein. J. Biol. Chem. 2008, 283, 15853–15860. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Piccoli, L.; Romeo, M.; Barzago, M.M.; Ravasio, S.; Foglierini, M.; Matkovic, M.; Sgrignani, J.; De Gasparo, R.; Prunotto, M.; et al. Machine learning analyses of antibody somatic mutations predict immunoglobulin light chain toxicity. Nat. Commun. 2021, 12, 3532. [Google Scholar] [CrossRef]
- Glenner, G.G. Amyloid deposits and amyloidosis. The beta-fibrilloses (first of two parts). N. Engl. J. Med. 1980, 302, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Sletten, K.; Natvig, J.B.; Husby, G.; Juul, J. The complete amino acid sequence of a prototype immunoglobulin-lambda light-chain-type amyloid-fibril protein AR. Biochem. J. 1981, 195, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Solomon, A.; Frangione, B.; Franklin, E.C. Bence Jones proteins and light chains of immunoglobulins. Preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J. Clin. Investig. 1982, 70, 453–460. [Google Scholar] [CrossRef]
- Annamalai, K.; Gührs, K.H.; Koehler, R.; Schmidt, M.; Michel, H.; Loos, C.; Gaffney, P.M.; Sigurdson, C.J.; Hegenbart, U.; Schönland, S.; et al. Polymorphism of Amyloid Fibrils In Vivo. Angew. Chem. Int. Ed. Engl. 2016, 55, 4822–4825. [Google Scholar] [CrossRef]
- Annamalai, K.; Liberta, F.; Vielberg, M.T.; Close, W.; Lilie, H.; Gührs, K.H.; Schierhorn, A.; Koehler, R.; Schmidt, A.; Haupt, C.; et al. Common Fibril Structures Imply Systemically Conserved Protein Misfolding Pathways In Vivo. Angew. Chem. Int. Ed. Engl. 2017, 56, 7510–7514. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.; Weiss, D.T.; Murphy, C.L.; Hrncic, R.; Wall, J.S.; Schell, M. Light chain-associated amyloid deposits comprised of a novel kappa constant domain. Proc. Natl. Acad. Sci. USA 1998, 95, 9547–9551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engvig, J.P.; Olsen, K.E.; Gislefoss, R.E.; Sletten, K.; Wahlström, O.; Westermark, P. Constant region of a kappa III immunoglobulin light chain as a major AL-amyloid protein. Scand. J. Immunol. 1998, 48, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Lavatelli, F.; Mazzini, G.; Ricagno, S.; Iavarone, F.; Rognoni, P.; Milani, P.; Nuvolone, M.; Swuec, P.; Caminito, S.; Tasaki, M.; et al. Mass spectrometry characterization of light chain fragmentation sites in cardiac AL amyloidosis: Insights into the timing of proteolysis. J. Biol. Chem. 2020, 295, 16572–16584. [Google Scholar] [CrossRef]
- Enqvist, S.; Sletten, K.; Westermark, P. Fibril protein fragmentation pattern in systemic AL-amyloidosis. J. Pathol. 2009, 219, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Kelly, J.W. The Kinetic Stability of a Full-Length Antibody Light Chain Dimer Determines whether Endoproteolysis Can Release Amyloidogenic Variable Domains. J. Mol. Biol. 2016, 428, 4280–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternke-Hoffmann, R.; Pauly, T.; Norrild, R.K.; Hansen, J.; Dupré, M.; Tucholski, F.; Duchateau, M.; Rey, M.; Metzger, S.; Boquoi, M.; et al. Universal amyloidogenicity of patient-derived immunoglobulin light chains. BioRxiv 2021. [Google Scholar] [CrossRef]
- Bansal, A.; Schmidt, M.; Rennegarbe, M.; Haupt, C.; Liberta, F.; Stecher, S.; Puscalau-Girtu, I.; Biedermann, A.; Fändrich, M. AA amyloid fibrils from diseased tissue are structurally different from in vitro formed SAA fibrils. Nat. Commun. 2021, 12, 1013. [Google Scholar] [CrossRef]
- Schönfelder, J.; Pfeiffer, P.B.; Pradhan, T.; Bijzet, J.; Hazenberg, B.P.C.; Schönland, S.O.; Hegenbart, U.; Reif, B.; Haupt, C.; Fändrich, M. Protease Resistance of ex vivo Amyloid Fibrils implies the proteolytic Selection of disease-associated Fibril Morphologies. BioRxiv 2021. [Google Scholar] [CrossRef]
- Kumar, S.; Murray, D.; Dasari, S.; Milani, P.; Barnidge, D.; Madden, B.; Kourelis, T.; Arendt, B.; Merlini, G.; Ramirez-Alvarado, M.; et al. Assay to rapidly screen for immunoglobulin light chain glycosylation: A potential path to earlier AL diagnosis for a subset of patients. Leukemia 2019, 33, 254–257. [Google Scholar] [CrossRef]
- Foss, G.S.; Nilsen, R.; Cornwell, G.C., III; Husby, G.; Sletten, K. A glycosylated Bence Jones protein and its autologous amyloid light chain containing potentially amyloidogenic residues. Scand. J. Immunol. 1998, 47, 348–354. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Larson, D.R.; Rajkumar, S.V.; Kyle, R.A.; Kumar, S.K.; Kourelis, T.; Arendt, B.; Willrich, M.; Dasari, S.; Murray, D. N-glycosylation of monoclonal light chains on routine MASS-FIX testing is a risk factor for MGUS progression. Leukemia 2020, 34, 2749–2753. [Google Scholar] [CrossRef]
- Mellors, P.W.; Dasari, S.; Kohlhagen, M.C.; Kourelis, T.; Go, R.S.; Muchtar, E.; Gertz, M.A.; Kumar, S.K.; Buadi, F.K.; Willrich, M.A.V.; et al. MASS-FIX for the detection of monoclonal proteins and light chain N-glycosylation in routine clinical practice: A cross-sectional study of 6315 patients. Blood Cancer J. 2021, 11, 50. [Google Scholar] [CrossRef]
- Prado, M.J.; Nicastri, A.L.; Costa, P.L.; Rockman, T.; Tersariol, I.L.; Nader, H.B.; Barros, R.T.; Prado, E.B. The renal and hepatic distribution of Bence Jones proteins depends on glycosylation: A scintigraphic study in rats. Braz. J. Med. Biol. Res. 1997, 30, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.; Wally, J.; Walsh, M.T.; Skinner, M.; Costello, C.E. Identification and location of a cysteinyl posttranslational modification in an amyloidogenic kappa1 light chain protein by electrospray ionization and matrix-assisted laser desorption/ionization mass spectrometry. Anal. Biochem. 2001, 295, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Klafki, H.W.; Kratzin, H.D.; Pick, A.I.; Eckart, K.; Karas, M.; Hilschmann, N. Complete amino acid sequence determinations demonstrate identity of the urinary Bence Jones protein (BJP-DIA) and the amyloid fibril protein (AL-DIA) in a case of AL-amyloidosis. Biochemistry 1992, 31, 3265–3272. [Google Scholar] [CrossRef]
- Omtvedt, L.A.; Husby, G.; Cornwell, G.G., 3rd; Kyle, R.A.; Sletten, K. The amino acid sequence of the glycosylated amyloid immunoglobulin light chain protein AL MS. Scand. J. Immunol. 1997, 45, 551–556. [Google Scholar] [CrossRef]
- Yazaki, M.; Liepnieks, J.J.; Callaghan, J.; Connolly, C.E.; Benson, M.D. Chemical characterization of a lambda I amyloid protein isolated from formalin-fixed and paraffin-embedded tissue sections. Amyloid 2004, 11, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Liu, H. Macro- and Micro-Heterogeneity of Natural and Recombinant IgG Antibodies. Antibodies 2019, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Klimtchuk, E.S.; Gursky, O.; Patel, R.S.; Laporte, K.L.; Connors, L.H.; Skinner, M.; Seldin, D.C. The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry 2010, 49, 9848–9857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Annamalai, K.; Schmidt, M.; Grigorieff, N.; Fändrich, M. Cryo-EM reveals the steric zipper structure of a light chain-derived amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, 6200–6205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piehl, D.W.; Blancas-Mejía, L.M.; Ramirez-Alvarado, M.; Rienstra, C.M. Solid-state NMR chemical shift assignments for AL-09 VL immunoglobulin light chain fibrils. Biomol. NMR Assign. 2017, 11, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Piehl, D.W.; Blancas-Mejía, L.M.; Wall, J.S.; Kennel, S.J.; Ramirez-Alvarado, M.; Rienstra, C.M. Immunoglobulin Light Chains Form an Extensive and Highly Ordered Fibril Involving the N- and C-Termini. ACS Omega 2017, 2, 712–720. [Google Scholar] [CrossRef]
- Close, W.; Neumann, M.; Schmidt, A.; Hora, M.; Annamalai, K.; Schmidt, M.; Reif, B.; Schmidt, V.; Grigorieff, N.; Fändrich, M. Physical basis of amyloid fibril polymorphism. Nat. Commun. 2018, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Hora, M.; Sarkar, R.; Morris, V.; Xue, K.; Prade, E.; Harding, E.; Buchner, J.; Reif, B. MAK33 antibody light chain amyloid fibrils are similar to oligomeric precursors. PLoS ONE 2017, 12, e0181799. [Google Scholar] [CrossRef]
- Lecoq, L.; Wiegand, T.; Rodriguez-Alvarez, F.J.; Cadalbert, R.; Herrera, G.A.; Del Pozo-Yauner, L.; Meier, B.H.; Böckmann, A. A Substantial Structural Conversion of the Native Monomer Leads to in-Register Parallel Amyloid Fibril Formation in Light-Chain Amyloidosis. Chembiochem 2019, 20, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Crowther, R.A.; Goedert, M.; Scheres, S.H. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. Elife 2019, 8, e43584. [Google Scholar] [CrossRef]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Wiese, S.; Adak, V.; Engler, J.; Agarwal, S.; Fritz, G.; Westermark, P.; Zacharias, M.; Fändrich, M. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 2019, 10, 5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Hughson, A.G.; Artikis, E.; Race, B.; Caughey, B. Structure of an infectious mammalian prion. BioRxiv 2021. [Google Scholar] [CrossRef]
- Radamaker, L.; Lin, Y.H.; Annamalai, K.; Huhn, S.; Hegenbart, U.; Schönland, S.O.; Fritz, G.; Schmidt, M.; Fändrich, M. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 2019, 10, 1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radamaker, L.; Baur, J.; Huhn, S.; Haupt, C.; Hegenbart, U.; Schönland, S.; Bansal, A.; Schmidt, M.; Fändrich, M. Cryo-EM reveals structural breaks in a patient-derived amyloid fibril from systemic AL amyloidosis. Nat. Commun. 2021, 12, 875. [Google Scholar] [CrossRef]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nokwe, C.N.; Hora, M.; Zacharias, M.; Yagi, H.; John, C.; Reif, B.; Goto, Y.; Buchner, J. The Antibody Light-Chain Linker Is Important for Domain Stability and Amyloid Formation. J. Mol. Biol. 2015, 427, 3572–3586. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haupt, C. The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure. Hemato 2021, 2, 505-514. https://doi.org/10.3390/hemato2030032
Haupt C. The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure. Hemato. 2021; 2(3):505-514. https://doi.org/10.3390/hemato2030032
Chicago/Turabian StyleHaupt, Christian. 2021. "The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure" Hemato 2, no. 3: 505-514. https://doi.org/10.3390/hemato2030032
APA StyleHaupt, C. (2021). The AL Amyloid Fibril: Looking for a Link between Fibril Formation and Structure. Hemato, 2(3), 505-514. https://doi.org/10.3390/hemato2030032