
Near-Infrared Transitions from the Singlet Excited States to the Ground Triplet State of the S2 Molecule

Abstract
:1. Introduction
2. Method of Calculations
3. Results for the S2 Molecule
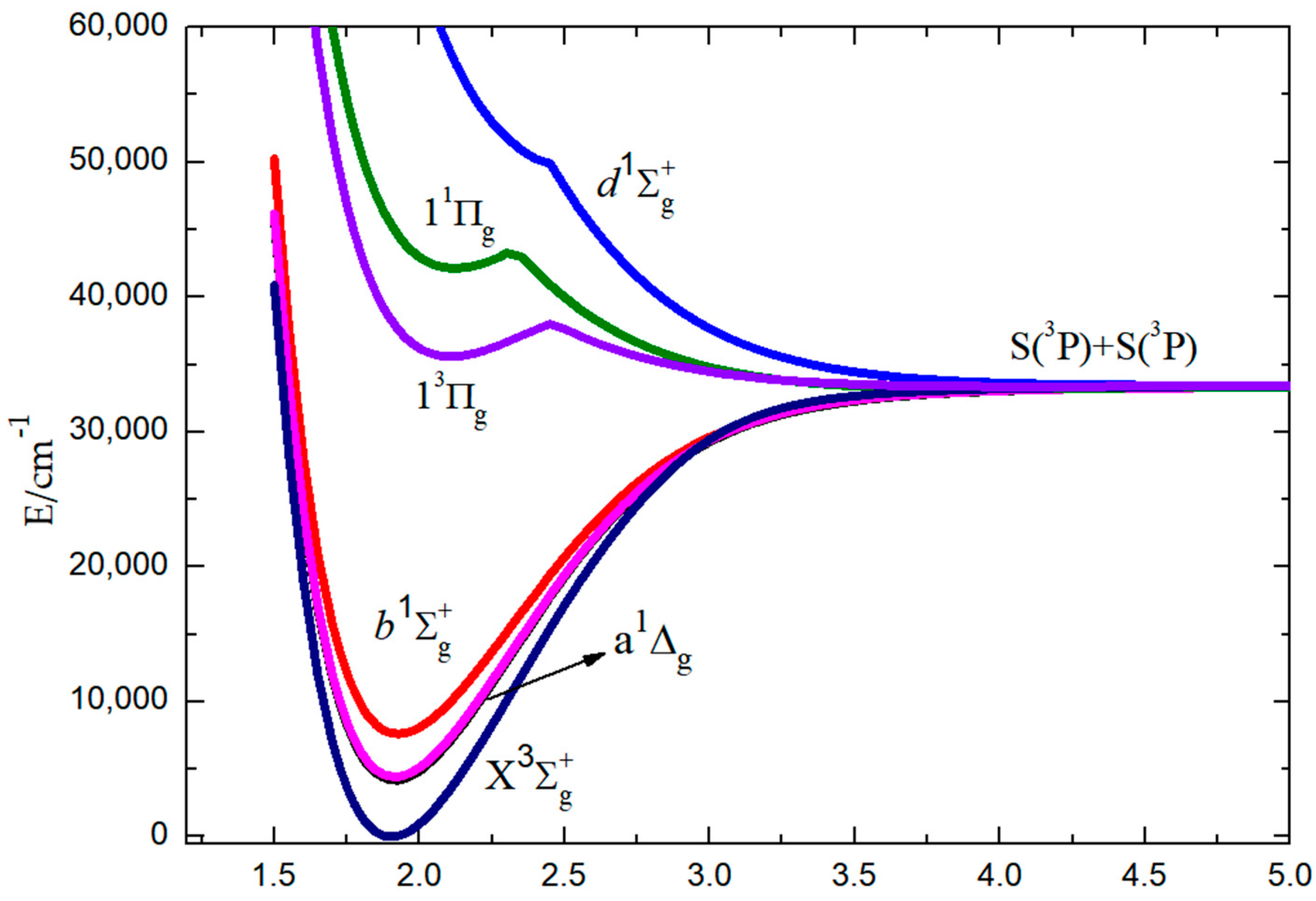
3.1. Energy Calculations in the S2 Spectrum
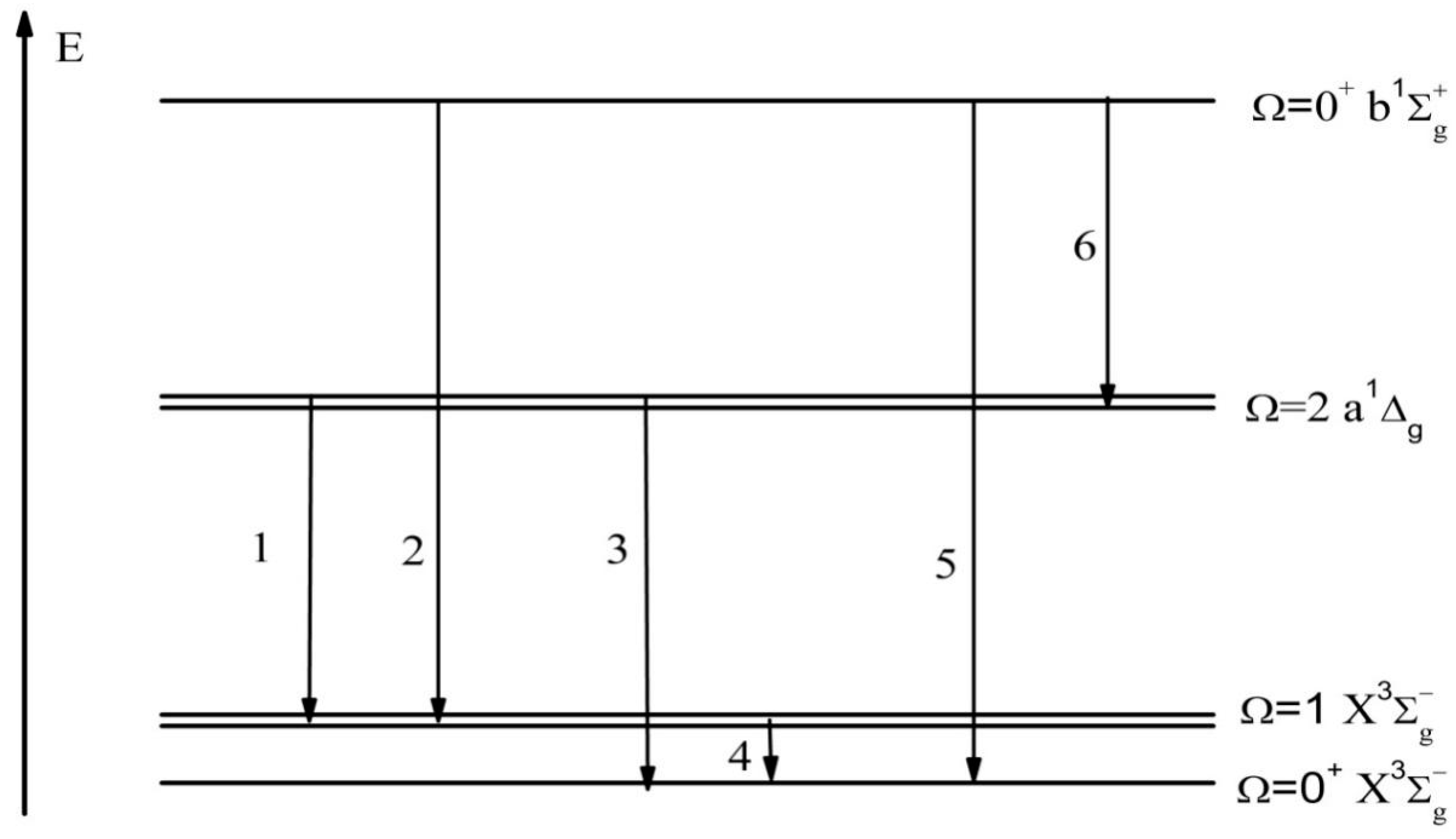
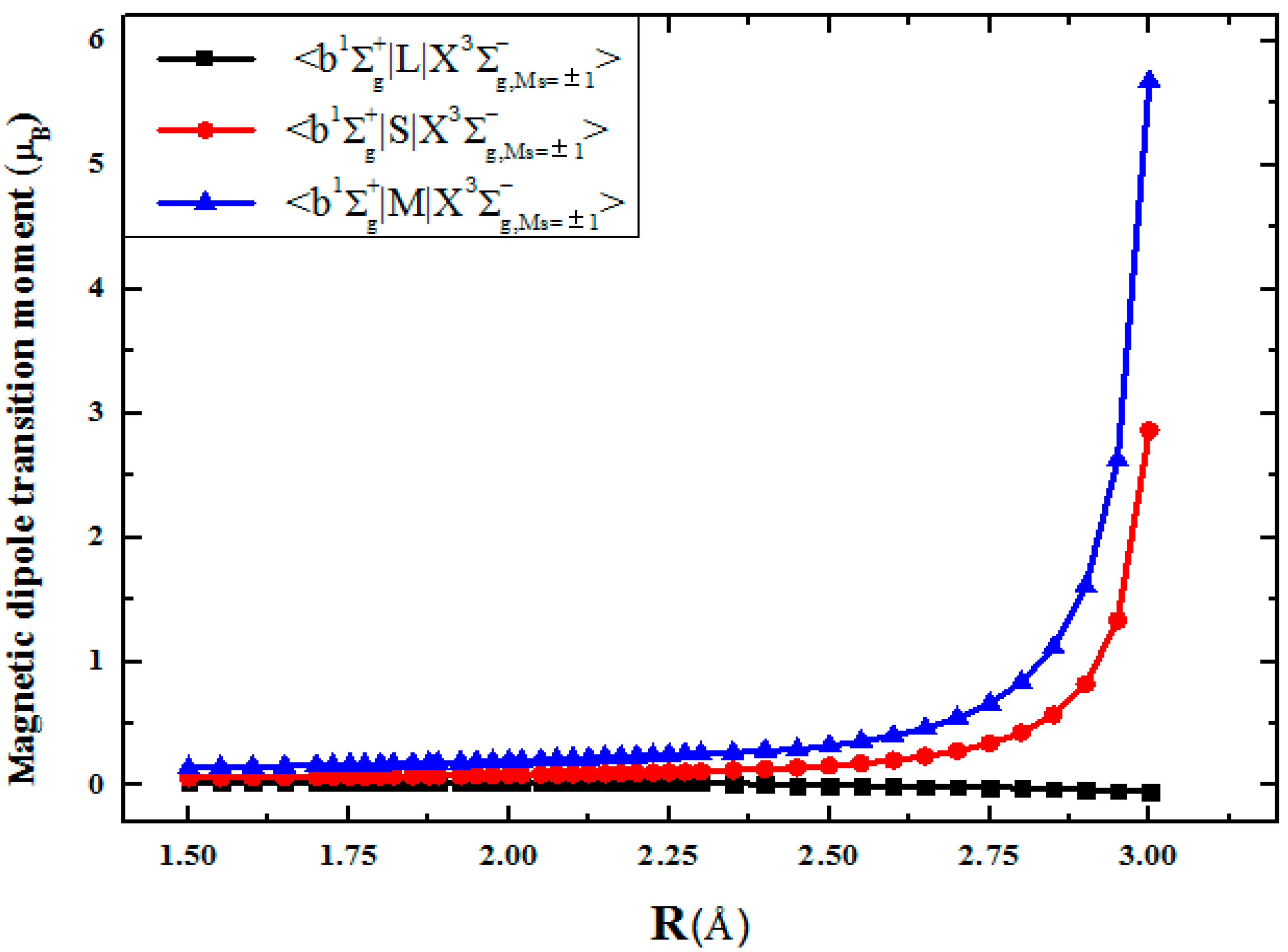
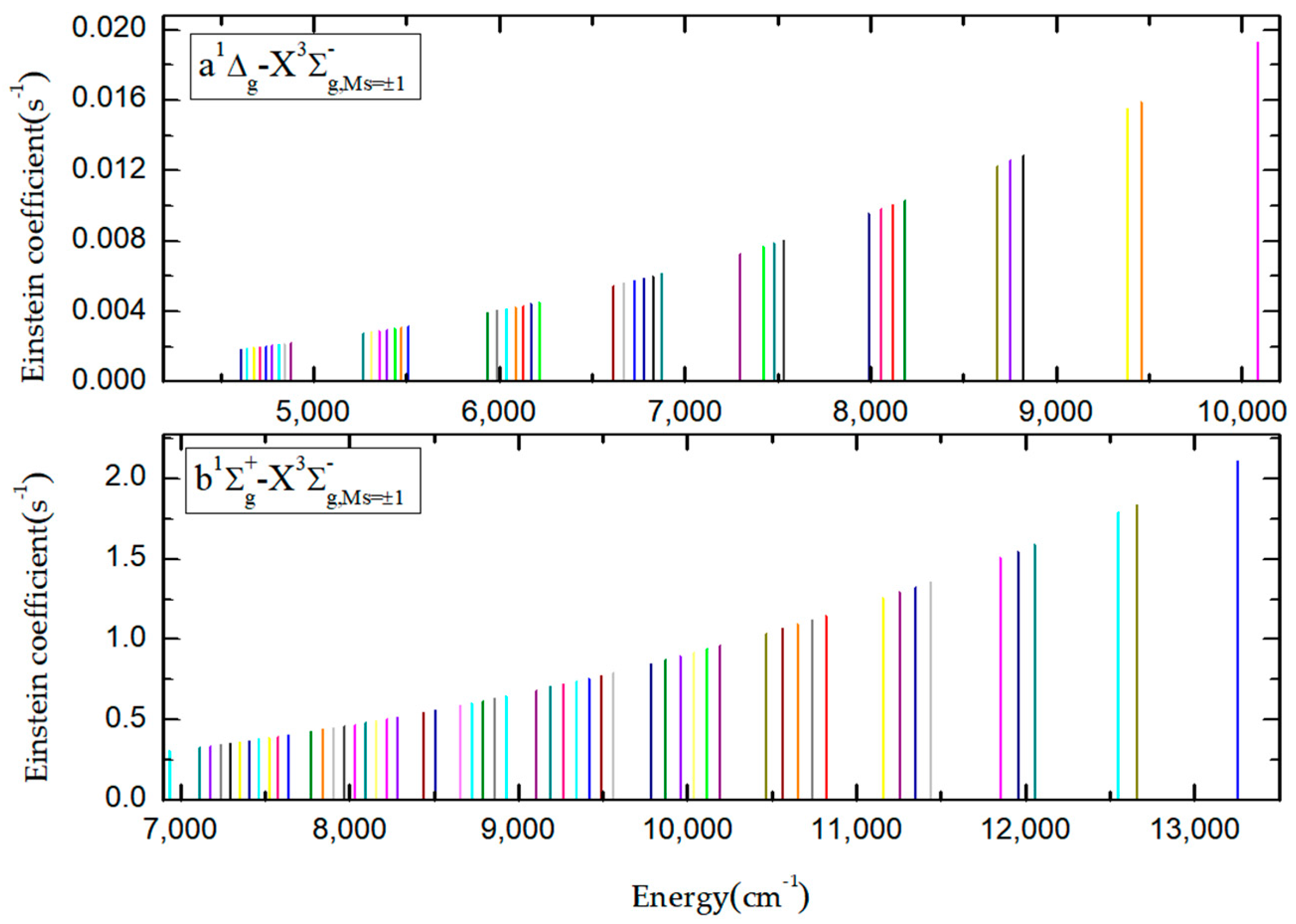
3.2. Magnetic Dipole Mechanisms for the Transitions in the S2 Spectrum
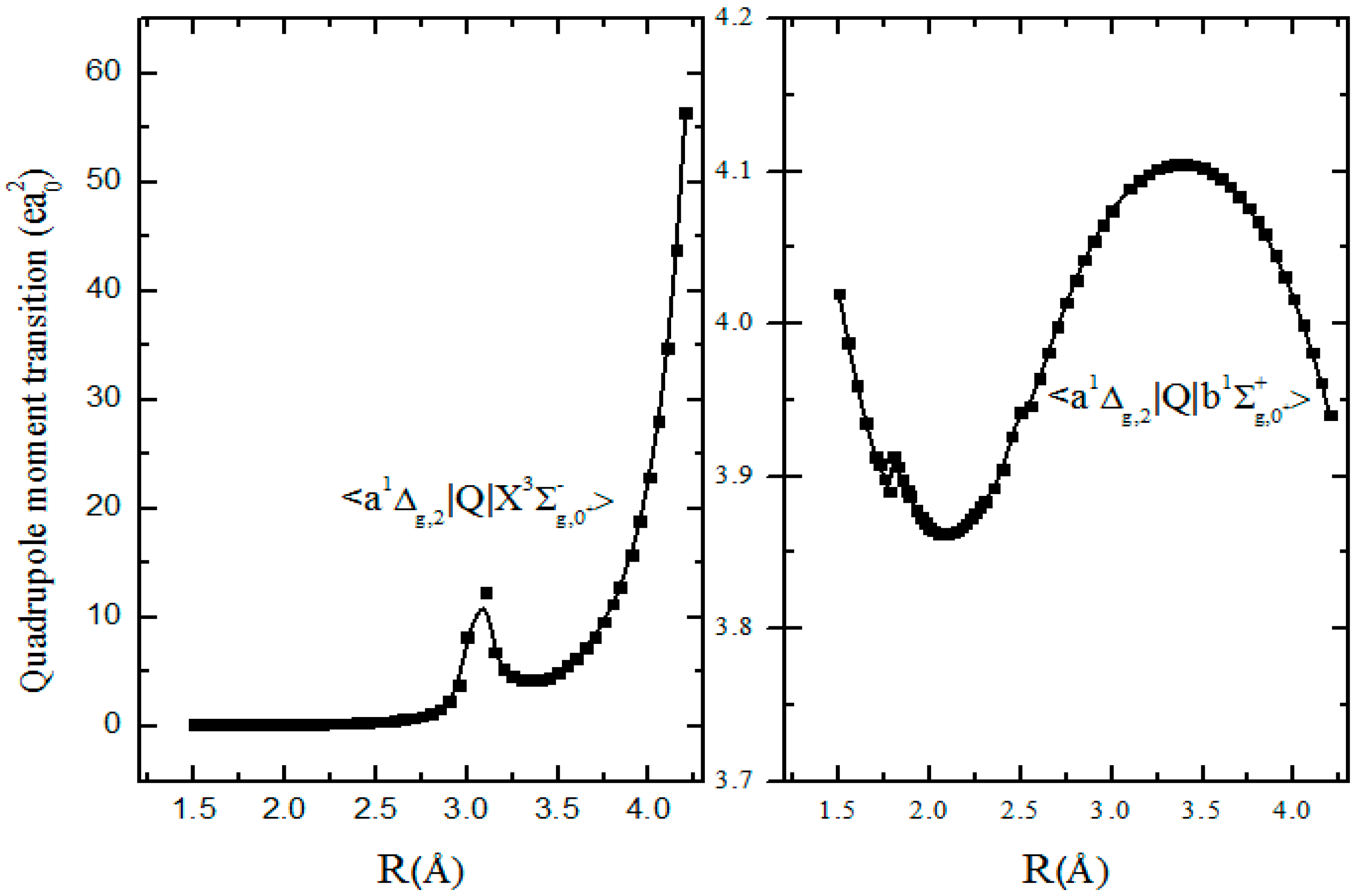
3.3. Electro-Quadrupole Mechanisms for Transitions between and States in the S2 Spectrum
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fowler, A.; Vaidya, W.M. The Spectrum of the Flame of Carbon Disulphide. Proc. R. Soc. London. Ser. A Contain. Pap. A Math. Phys. Character 1931, 132, 310–330. [Google Scholar]
- Tanaka, Y.; Ogawa, M. Emission spectrum of S2 in the vacuum ultraviolet region. J. Chem. Phys. 1962, 36, 726–730. [Google Scholar] [CrossRef]
- Maeder, R.; Miescher, E. Absorption band spectrum of S2 in the Schumann region. Nature 1948, 161, 393. [Google Scholar] [CrossRef] [PubMed]
- Bondybey, V.E.; English, J.H. B3Σ-u predissociation and relaxation processes in matrix isolated S2. J. Chem. Phys. 1979, 72, 3113–3122. [Google Scholar] [CrossRef]
- Yuan, D.F.; Trabelsi, T.; Zhang, Y.R.; Francisco, J.S.; Wang, L.S. Probing the Electronic Structure and Bond Dissociation of SO3 and SO3– Using High-Resolution Cryogenic Photoelectron Imaging. J. Am. Chem. Soc. 2022, 144, 13740–13747. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.R.; Rama, G.K.; Nazeer, A.Y.; Baba, B.D.; Narasimhulu, K.; Siva Sankar Reddy, L. Spectroscopic studies of molecules observed in comets. Indian J. Pure Ap. Phy. 2005, 43, 237–245. [Google Scholar]
- De Almeida, A.A.; Singh, P.D. Photodissociation lifetime of 32S2 molecule in comets. Earth Moon Planets 1986, 36, 117–125. [Google Scholar] [CrossRef]
- Spencer, J.R.; Jessup, K.L.; McGrath, M.A.; Ballester, G.E.; Yelle, R. Discovery of gaseous S2 in Io’s Pele plume. Science 2000, 288, 1208–1210. [Google Scholar] [CrossRef]
- Xue, J.L.; Yuan, X.; Li, R.; Liu, X.S.; Yan, B. Theoretical study on predissociation of B3Σ-u of sulfur dimer. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 241, 118679. [Google Scholar] [CrossRef]
- Kiljunen, T.; Eloranta, J.; Kunttu, H.; Khriachtchev, L.; Pettersson, M.; Räsänen, M. Electronic structure and short-range recombination dynamics of S2 in solid argon. J. Chem. Phys. 2000, 112, 7475–7483. [Google Scholar] [CrossRef]
- Frederix, P.W.J.M.; Yang, C.H.; Groenenboom, G.C.; Parker, D.H.; Alnama, K.; Western, C.M. Photodissociation imaging of diatomic sulfur (S2). J. Phys. Chem. A 2009, 113, 14995–15005. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Fujihara, K.; Takahashi, O.; Kohguchi, H.; Yamasaki, K. Kinetics and dynamics on the formation of S2(X3Σ−g−a1Δg) in the S(1D) + OCS reaction. J. Phys. Chem. A 2014, 118, 9330–9337. [Google Scholar] [CrossRef] [PubMed]
- Green, M.E.; Western, C.M. Upper vibrational states of the B″3Πu state of 32S2. J. Chem. Soc. Faraday Trans. 1997, 93, 365–372. [Google Scholar] [CrossRef]
- Minaev, B.F.; Minaeva, V.A. MCSCF response calculations of the excited states properties of the O2 molecule and a part of its spectrum. Phys. Chem. Chem. Phys. 2001, 3, 720–729. [Google Scholar] [CrossRef]
- Wieland, K. Thermo-optical dissociation of sulphur dioxide. Trans. Faraday Soc. 1934, 30, 260–265. [Google Scholar] [CrossRef]
- Donovan, R.J.; Husain, D.; Jackson, P.T. Transient species in the photolysis of sulphur monochloride, including S2 (a1Δg). Trans. Faraday Soc. 1968, 64, 1798–1805. [Google Scholar] [CrossRef]
- Chiang, S.Y.; Lee, Y.P. Red and near-infrared laser-induced emission of S2 in an Ar matrix. J. Chem. Phys. 1988, 89, 13–19. [Google Scholar] [CrossRef]
- Minaev, B.F. Intensity of singlet-triplet transitions in the oxygen molecule and the selective effect of an external heavy atom. Opt. Spectrosc. 1978, 45, 1202–1207. [Google Scholar]
- Minaev, B.F. Electronic mechanisms of activation of molecular oxygen. Russ. Chem. Rev. 2007, 76, 1059. [Google Scholar] [CrossRef]
- HESS, B.A.; Buenrer, R.J. Ab initio calculation of the Zero-Field splittiongs of the X3Σ-g, B3Πg,1 states of the S2 molecule. Chem. Phys. 1982, 71, 79–85. [Google Scholar] [CrossRef]
- Klotz, R.; Marian, C.M.; Peyerimhoff, S.D. Calculation of spin-forbidden radiative transitions using correlated wavefunctions lifetimes of b1Σ+, a1Δ states in O2, S2 and SO. Chem. Phys. 1984, 89, 223–236. [Google Scholar] [CrossRef]
- Fink, E.H.; Kruse, H.; Ramsay, D.A. The high-resolution emission spectrum of S2 in the near infrared, The b1Σ+g−X3Σ−g system. J. Mol. Spectrosc. 1986, 119, 377–387. [Google Scholar] [CrossRef]
- Setzer, K.D.; Kalb, M.; Fink, E.H. The a1Δg-X3Σ-g magnetic dipole transition of S2. J. Mol. Spectrosc. 2003, 221, 127–130. [Google Scholar] [CrossRef]
- Barnes, I.; Becker, K.H.; Fink, E.H. Near-infrared emissions from the 1Δg and 1Σ+g states of S2. Chem. Phys. Lett. 1979, 67, 314–317. [Google Scholar]
- Fink, E.H.; Kruse, H.; Setzer, K.D. High resolution fourier-transform spectra of the a1Δg−X3Σ−g, b1Σ+g−X3Σ−g and b1Σ+g−a1Δg systems of O2, SO, S2 and isoelectronic molecules in the nir region. Acta Phys. Hung. 1990, 67, 67–72. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. W. I. Res. Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- A’Hearn, M.F.; Feldman, P.D.; Schleicher, D.G. The discovery of S2 in comet IRAS-Araki-Alcock 1983d. Astrophys. J. 1983, 274, L99–L103. [Google Scholar] [CrossRef]
- Epler, J.E.; Verdeyen, J.T. Broad-Band Gain in Optically Pumped S2. IEEE J. Quantum Elect. 1983, 19, 1686–1691. [Google Scholar] [CrossRef]
- Glassgold, A.E. Circumstellar photochemistry. Annu. Rev. Astron. Astr. 1996, 34, 241–277. [Google Scholar] [CrossRef]
- Grim, R.J.A.; Greenberg, J.M. Photoprocessing of H2S in interstellar grain mantles as an explanation for S2 in comets. Astron. Astrophys. 1987, 181, 155–168. [Google Scholar]
- Kim, S.J.; A’Hearn, M.F.; Larson, S.M. Multi-cycle fluorescence, Application to S2 in comet IRAS-Araki-Alcock 1983VII. Icarus 1990, 87, 440–451. [Google Scholar] [CrossRef]
- Lewis, J.S.; Kreimendahl, F.A. Oxidation state of the atmosphere and crust of Venus from pioneer Venus results. Icarus 1980, 42, 330–337. [Google Scholar] [CrossRef]
- Liszt, H.S. Upper limits on the abundance of the sulfur dimer in molecular clouds. Astrophys J. 1978, 219, 454–457. [Google Scholar] [CrossRef]
- Mitchell, G.F. Effects of shocks on the sulfur chemistry of a dense interstellar cloud. Astrophys. J. 1984, 287, 665–670. [Google Scholar] [CrossRef]
- Noll, K.S.; McGrath, M.A.; Trafton, L.M.; Atreya, S.K.; Caldwell, J.J.; Weaver, H.A. HST spectroscopic observations of Jupiter after the collision of comet Shoemaker—Levy 9. Science 1995, 267, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Shnitko, I.; Fulara, J.; Garkusha, I.; Nagy, A.; Maier, J.P. Electronic transitions of S2− and S3− in neon matrixes. Chem. Phys. 2008, 346, 8–12. [Google Scholar]
- Huber, K.P.; Herzberg, G. Constants of Diatomic Molecules; Van Nostrand Reinhold Co.: New York, NY, USA, 1979. [Google Scholar]
- Swope, W.C.; Lee, Y.P.; Schaefer, H.F., III. Diatomic sulfur: Low lying bound molecular electronic states of S2. J. Chem. Phys. 1979, 70, 947–953. [Google Scholar] [CrossRef]
- Bielefeld, M.; Elfers, G.; Fink, E.H.; Kruse, H.; Wildt, J.; Winter, R. O2(a1Δg)-sensitized chemiluminescence of a1Δ → X3Σ− and b1Σ+ → X3Σ− transitions of group VI—Group VI and group V–group VII diatomic molecules. J. Photoch. 1984, 25, 419–438. [Google Scholar] [CrossRef]
- Veillard, A. Gaussian basis set for molecular wavefunctions containing second-row atoms. Theo. Chim. Acta 1968, 12, 405–411. [Google Scholar] [CrossRef]
- Fink, E.H.; Setzer, K.D.; Kottsieper, U.; Ramsay, D.A.; Vervloet, M. The a1Δ (a2)—X3Σ-(X21) electronic band system of selenium monoxide. J. Mol. Spectrosc. 1988, 131, 127–132. [Google Scholar] [CrossRef]
- Minaev, B.F.; Murugan, N.A.; Ågren, H. Dioxygen spectra and bio-activation. Int. J Quant. Chem. 2013, 113, 1847–1867. [Google Scholar] [CrossRef]
- Sveshnikova, E.B.; Minaev, B.F. Mechanism of the nonradiative 1Δg−3Σg− transition in molecular oxygen in solution. Opt. Spectrosc. 1983, 54, 320–322. [Google Scholar]
- Gordon, I.E.; Kassi, S.; Campargue, A.; Toon, G.C. First identification of the a1Δg-X3Σ-g electric quadrupole transitions of oxygen in solar and laboratory spectra. J. Quant Spectrosc. Radiat. Transf. 2010, 111, 1174. [Google Scholar] [CrossRef]
- Minaev, B.F. Effect of spin-orbit coupling on the intensity of magnetic dipole transitions in molecular oxygen. Sov. Phys. J. 1978, 21, 1205–1209, Erratum in Izv. Vyssh. Uchebn. Zaved. Fiz. 1978, 9, 115. [Google Scholar] [CrossRef]
- Minaev, B.F. Spin-orbit coupling mechanism of singlet oxygen a1Δ g quenching by solvent vibrations. Chem. Phys. 2017, 483, 84–95. [Google Scholar] [CrossRef]
- Xiao, L.D.; Xue, J.L.; Liu, Y.; Yan, B.; Minaev, B.F. Calculation of the singlet-triplet magnetic and electro-quadrupole transitions intensity for Ge2 molecule. Mol. Phys. 2020, 120, e20745622020. [Google Scholar] [CrossRef]
- Minaev, B.F.; Panchenko, O.O.; Minaeva, V.A.; Agren, H. Triplet state harvesting and search for forbidden transition intensity in the nitrogen molecule. Front. Chem. 2022, 10, 1005684. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Te/cm−1 | Re/Å | ωe/cm−1 | ωeχe/cm−1 | Be/cm−1 | |
|---|---|---|---|---|---|
| 0 | 1.9027 | 709.6586 | 2.8198 | 0.2913 | |
| 1.889 Expt [37] | 725.65 Expt [37] | ||||
| 1.90 [38] | 760 [38] | ||||
| 1.8960 [9] | 729 [9] | 3.2096 [9] | 0.2933 [9] | ||
| 4459.3688 | 1.9150 | 682.6867 | 3.0585 | 0.2876 | |
| 1.898 Expt [37] | 702.35 Expt [37] | ||||
| 1.907 [38] | 746 [38] | ||||
| 4390 [9] | 1.9058 [9] | 703 [9] | 3.2536 [9] | 0.2903 [9] | |
| 7649.5722 | 1.9249 | 650.8639 | 2.8708 | 0.2843 | |
| 669.7 Expt [37] | |||||
| 1.914 [38] | 732 [38] | ||||
| 7675 [9] | 1.9181 [9] | 674 [9] | 3.3856 [9] | 0.2866 [9] | |
| 35,619.7022 | 2.1081 | 435.4006 | 7.7393 | 0.2373 | |
| 42,170.9966 | 2.1213 | 459.4193 | 6.7306 | 0.2342 |
| Basis | Spin-Orbit Matrix Elements with the State for Other States (cm−1) | Spin-Orbit Matrix Elements with the State for Other States (cm−1) | |||
|---|---|---|---|---|---|
| best CBS | 429.85 ± 0.17 | 215.90 ± 0.06 | −217.38 ± 0.01 | −212.76 ± 0.02 | 201.47 ± 0.00 |
| basis D [20,41] | 422.5 | 200.3 | −203.1 | ||
| basis D1 (core2) [20] | 423.0 | 202.7 | −204.0 | ||
| basis E [20] | 410.3 | 200.9 | −200.8 | ||
| basis F [20] | 416.6 | 203.9 | −204.9 | ||
| basis G (core2) [21] | −200.2 | 188.6 | |||
| Rs-s (Å) | |||
| 1.8 | 1.23995 | 1.23995 | 1.19769 |
| 1.85 | 1.25557 | 1.25557 | 1.20622 |
| 1.899 | 1.25986/1.26317 [21] | 1.25986/1.26317 [21] | 1.21257/1.22810 [21] |
| 1.95 | 1.25704 | 1.25704 | 1.21662 |
| 2.0 | 1.25890 | 1.25890 | 1.21828 |
| 2.05 | 1.25894 | 1.25894 | 1.21742 |
| 2.1 | 1.25751 | 1.25751 | 1.21392 |
| Rs-s (Å) | |||
| 1.8 | 1.21115 | 1.21115 | 1.19769 |
| 1.85 | 1.21464 | 1.21464 | 1.20622 |
| 1.899 | 1.21624/1.26317 [21] | 1.21624/1.26317 [21] | 1.21257/1.22810 [21] |
| 1.95 | 1.21697 | 1.21697 | 1.21662 |
| 2.0 | 1.21202 | 1.21202 | 1.21828 |
| 2.05 | 1.20255 | 1.20255 | 1.21742 |
| 2.1 | 1.18724 | 1.18724 | 1.21392 |
| R(Å) | (i a.u.) | (i a.u.) | (iμB) | (i a.u.) | (iμB) |
|---|---|---|---|---|---|
| 1.5 | 0.020765 | 0.060717 | 0.142199 | 0.020114 | 0.020114 |
| 1.7 | 0.024100 | 0.068487 | 0.161075 | 0.023092 | 0.023092 |
| 1.752 | 0.025084 | 0.070752 | 0.166587 | 0.023996 | 0.023996 |
| 1.800 | 0.025882 | 0.073154 | 0.172189 | 0.024779 | 0.024779 |
| 1.82 | 0.026272 | 0.074146 | 0.174564 | 0.025143 | 0.025143 |
| 1.889 | 0.027554 | 0.077828 | 0.183211 | 0.026369 | 0.026369 |
| 1.95 | 0.028579 | 0.080969 | 0.190517 | 0.027407 | 0.027407 |
| 2 | 0.029324 | 0.084301 | 0.197926 | 0.028197 | 0.028197 |
| 2.02 | 0.029593 | 0.085734 | 0.201062 | 0.028492 | 0.028492 |
| 2.1 | 0.030449 | 0.092158 | 0.214766 | 0.029496 | 0.029496 |
| 2.15 | 0.030757 | 0.096839 | 0.224436 | 0.029902 | 0.029902 |
| 2.2 | 0.030805 | 0.102173 | 0.235151 | 0.029974 | 0.029974 |
| 2.4 | 0.012961 | 0.133883 | 0.280727 | 0.020556 | 0.020556 |
| 2.6 | −0.004597 | 0.202326 | 0.400056 | −0.005300 | −0.005300 |
| 2.8 | −0.019259 | 0.426683 | 0.834106 | −0.024475 | −0.024475 |
| 3 | −0.050699 | 2.861899 | 5.673100 | −0.072389 | −0.072389 |
| Ω-Ω Transition | ∆E/cm−1 | A/s−1 | λ/nm | FCF | τ/s |
|---|---|---|---|---|---|
| − | 7641 | 0.403824 | 1308.94 | 0.9100 | 2.4763 |
| 7961 Expt [24]/7981 Expt [39] | |||||
| 8970 [21] a | 0.423 [21] a /0.3 [21] b | 2.4 [21] a/3.4 [21] b | |||
| − | 4197 | 0.001386 | 2382.95 | 0.9702 | 721.2904 |
| 5551 [21] a | 0.00286 [21] | 350 [21] a | |||
| 4700 Expt [24] | |||||
| − | 3443 | 0.001228 | 2904.68 | 0.9820 | 814.1196 |
| 3419 [21] a/3617 [21] c/3586 [5,39] d | 960 [21] a/745 [21] c/780 [21] d | ||||
| − | 4221 | 0.000010 | 2369.65 | 0.9705 | 1618.42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, L.; Yan, B.; Minaev, B.F. Near-Infrared Transitions from the Singlet Excited States to the Ground Triplet State of the S2 Molecule. Physchem 2023, 3, 110-124. https://doi.org/10.3390/physchem3010009
Xiao L, Yan B, Minaev BF. Near-Infrared Transitions from the Singlet Excited States to the Ground Triplet State of the S2 Molecule. Physchem. 2023; 3(1):110-124. https://doi.org/10.3390/physchem3010009
Chicago/Turabian StyleXiao, Lidan, Bing Yan, and Boris F. Minaev. 2023. "Near-Infrared Transitions from the Singlet Excited States to the Ground Triplet State of the S2 Molecule" Physchem 3, no. 1: 110-124. https://doi.org/10.3390/physchem3010009
APA StyleXiao, L., Yan, B., & Minaev, B. F. (2023). Near-Infrared Transitions from the Singlet Excited States to the Ground Triplet State of the S2 Molecule. Physchem, 3(1), 110-124. https://doi.org/10.3390/physchem3010009